
BY: SAI MANOGNA (MSIWM014)
Paul Ehrlich described the process of tolerance and autoimmunity. He further realized that the immune system could be incorrect and could concentrate its invasion on self-antigens instead of responding only to foreign antigens. A variety of chronic and acute illness, including multiple sclerosis, rheumatoid arthritis, lupus erythematosus, and certain forms of diabetes, may result from this disease, which he called ‘horror autotoxicus.’ Simply put, these diseases arise from the host’s humoral and cellular immune systems failing to differentiate self from non-self, resulting in autoantibodies and self-reactive T cells attacking self-cells and organs.
Tolerance: To protect a person from potentially self-reactive lymphocytes, various mechanisms exist; these are given the general term tolerance.
Central tolerance: Until the cells are allowed to mature, a primary mechanism called central tolerance deletes T or B cell clones if they have receptors that recognize self-antigens with more than a low threshold affinity. In the main lymphoid glands, bone marrow, and thymus, core tolerance takes place. There are additional precautions to restrict their operation since central tolerance is not complete, and some of the self-reactive lymphocytes find their way into the secondary lymphoid tissues.

Peripheral tolerance: Peripheral tolerance allows lymphocytes in secondary lymphoid tissues inactive or anergic, requires these backup safeguards. The lifetime of activated lymphocytes regulated by programs that cause cell death ( apoptosis) upon receipt of signals further limits the likelihood of damage from self-reactive lymphocytes.
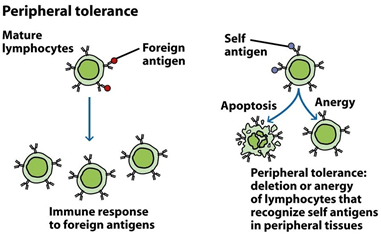
Autoimmunity: T or B cells’ self-reactive clones are sometimes activated, producing humoral or cell-mediated responses to self-antigens, despite this layered regulation system. Such an inadequate immune system response to self-components is referred to as autoimmunity. Autoimmune reactions, often with fatal consequences, may cause severe cell and organ damage.
Establishment and maintenance of tolerance:
To avoid the reaction of its cells and antibodies with host components and the onset of autoimmune disease, many layers of defense exerted by immune system are known under general heading of tolerance, which is defined as a state of unresponsiveness to an antigen. The mechanisms that mediate this unresponsiveness will differ. Finding an antigen in the immune system leads to an immune response under normal circumstances, but presenting the antigen in some alternative form can lead to tolerance or non-responsiveness.
Instead of immunogens, antigens that induce tolerance are known to be, depending on how it is addressed to the immune system, the same chemical compound may be both an immunogen and a tolerogen. For instance, an antigen presented to T cells without adequate costimulation results in a type of tolerance known as anergy, while a potent immunogen may become the same antigen presented with costimulatory molecules present.

Factors that encourage tolerance rather than activation by a given antigen of the immune system include the following:
1. High antigen doses
2. Antigen persistence in the host
3. Intravenous presentation or oral presentation
4. Lack of Adjuvants
5. Low concentrations of costimulators
Mechanism of Central tolerance:
The dominant mechanism for preserving tolerance is deletion during early maturation of lymphocyte clones that can react with self-components. Consider the pathways in T-cell or B-cell receptors that produce diversity. The genetic rearrangements that give rise to a functional TCR or Ig occur through a mechanism through which any gene in the V-region can relate to any portion of the D or J gene. It implies that it is theoretically possible to produce V regions reacting with self-antigen. If this were permitted to occur frequently, mature functional T or B cells could be formed by certain TCR or Ig receptors that recognize self and follow autoimmune disease. Alternatively, this so-called central tolerance mechanism acts to suppress autoreactive B cells in the bone marrow and autoreactive T cells in the thymus by modifying or editing the receptors on those clones that respond with themselves, reducing their affinity for self-antigens below a critical threshold that would lead to disease. Although our understanding of the specific molecular mechanisms mediating central tolerance in T and B cells is not complete, it is known that B and T cells are subjected to a developmentally mediated event known as negative selection, leading to death in cells carrying potentially autoreactive receptors of TCR or Ig.
1. The elegant work of C. Goodnow and colleagues is one of the classic experiments showing that self-reactive lymphocytes are eliminated or inactivated after an encounter with self-antigen.
2. Mice expressing hen egg-white lysozyme (HEL) specific transgenic immunoglobulin were mated to transgenic mice expressing HEL B cells formed in the offspring encountered HEL in the bone marrow when anti-HEL * mice were mated to HEL animals.
3. They noticed that mature B cells expressing anti-HEL were absent from the F1 mice.
4. Subsequent studies have shown that autoreactive evolving B cells, for the most part, are deleted by apoptosis induction in the bone marrow.
5. These findings were expanded by David Nemazee and colleagues and demonstrated that some developing B cells would undergo a process called receptor editing.
6. The antigen-specific V region is edited in cells undergoing a different V-region gene segment swapped via V(D)J recombination for the autoreactive V gene segment (Editing occurs most often within the VL, rather than the VH region.)
7. Receptor editing and clonal deletion or apoptosis are believed to be one of the pathways leading to core deletion.
8. In growing B cells, tolerance. T cells growing in the thymus with too high an affinity for self-antigens are deleted, mainly through the induction of apoptosis and related mechanisms.

Mechanism of Peripheral tolerance:
Another important finding from the anti-HEL transgenic experiments was that it caused the clonal deletion of all immature B cells with anti-HEL Ig if HEL was expressed on the cells’ membrane. However, the B cells matured and exited the bone marrow and were located in the periphery when HEL was secreted and detected as a soluble protein. However, these cells were not receptive to the antigen HEL and lived in a condition known as anergy. Anergy can be characterized as a lack of antigenic stimulus responsiveness. As the HEL studies and numerous other examples demonstrate, central tolerance is not a fool proof approach.
It does not entirely eradicate all potential self-reactive lymphocytes since
(a) not all self-antigens are expressed in the central lymphoid organs where there is a negative selection, and
(b) there is a negative selection, until the clonal deletion is activated, the threshold requirement for self-antigens’ affinity enables some weakly self-reactive clones to survive the weeding-out process.
1. In certain instances, in the thymus or bone marrow, self-reactive T or B cells can escape deletion and appear in the periphery.
2. These cells are inactivated by a type of tolerance termed peripheral tolerance. Peripheral tolerance can be characterized as the inactivation of the periphery’s self-reactive T cells or B cells, making them unable to respond to themselves.
3. As with central tolerance, before the requisite experimental proof, peripheral tolerance was expected.
4. The HEL provided to mature anti-HEL B cells in peripheral tissues inactivates these B cells, and they never migrate to lymphoid follicles in the spleen or lymph node, using the expressing mouse models mentioned above.
5. Note that B cells in lymphoid follicles / germinal centers become antibody-secreting plasma cells after maturation and selection.
6. In these experiments, a significant aspect to appreciate is that T cells that recognize HEL in the HEL-expressing mouse are deleted before maturation and release because of central tolerance, the antigen can be recognized by the B cells, but T cells do not have any subsequent support.
7. These experiments showed that when mature B cells encounter soluble antigen, they become unresponsive, OFF, in the absence of T-cell assistance and never move to germinal centers and become anergic.
8. Early experiments carried out by M. K. Jenkins, D. Mueller, and RH. Schwartz demonstrated that CD4 + T-cell clones were unresponsive in vitro when stimulated solely via the TCR; to characterize this state of unresponsiveness, they used the term clonal anergy.
9. Subsequent evidence from several laboratories showed that the antigen-presenting cell interaction between CD28 on the T cell and B7 provided the requisite costimulatory signals needed to activate T cells.
10. A detailed review of costimulation, revealing the presence of inhibitory receptors such as CTLA-4, culminated in the understanding that CD28 / B7 signals provide necessary costimulatory signals for T-cell activation.
11. CTLA-4 binds to B7, like CD28, but CTLA-4 inhibits T-cell activation instead of supplying activating signals.
12. After T cells are activated, CTLA-4 expression is induced, maintaining control and regulation of T cells’ activation.
13. When the gene encoding this molecule was deleted, the role of CTLA-4 in tolerance was appreciated. Mice were missing CTLA-4 show massive lymphocyte proliferation and autoimmune disease, indicating that this molecule plays an essential role in maintaining peripheral tolerance.

Regulatory T cells:
1. Regulatory T (Treg cells) cells can also induce Peripheral Tolerance. Acting in and at secondary lymphoid tissues, Treg cells downregulate autoimmune processes due to inflammation.
2. Recall that Treg cells are a unique subset of high levels of the IL-2R alpha chain (CD25) expressed by CD4 + T cells; it has been shown that Treg cells originate from a subset of T cells that express receptors in the thymus with intermediate affinity for self-antigens.
3. Some of these cells up-regulate the Foxp3 and transcription factor and then grow into Treg cells that are capable of suppressing self-antigen reactions.
4. Early studies in non-obese diabetic (NOD) mice and BB rats (the first animal model of spontaneous autoimmune type I diabetes), two strains vulnerable to the development of autoimmune-based diabetes, demonstrated the ability of Treg cells to suppress an immune response.
5. When these animals are infused with normal CD4 + T cells by histocompatible donors, the development of diabetes in NOD mice and BB rats is delayed.
6. Further characterization of CD4 + T cells showed that a subset noteworthy for expressing elevated CD25 levels was responsible for the suppression of diabetes in these animal models.
7. The mechanisms by which Treg cells suppress immune responses are an intensive study area, but it is clear that suppression is regulated, at least in part, by productive cytokines, including IL-10 and TGFβ.

In preserving both central and peripheral tolerance, cell death plays a significant role. It is demonstrated by the growth of systemic autoimmune diseases in either the death receptor, Fas, or Fas ligand (FasL) mutations in mice that occur naturally. Increased Fas and FasL levels are expressed by activated T cells. The presence of Fas by FasL in both B and T cells induces a rapid apoptotic death known as activation-induced cell death (AICD). Mice carrying Fas (LPR / LPR) or FasL (gold / gold) inactivating mutations cannot participate in the AICD pathway and develop the autoimmune disease early in life.
