BY: RAHUL ANDHARIA (MSIWM001)
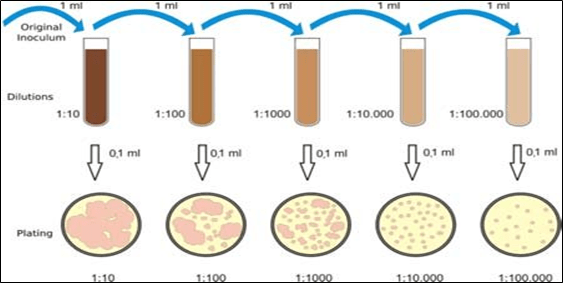
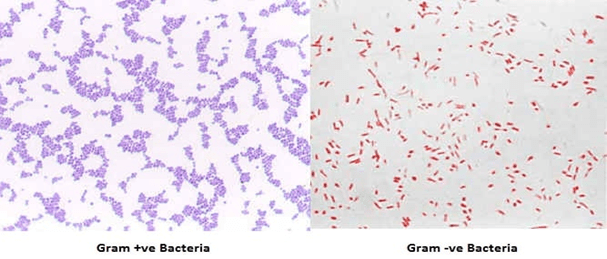
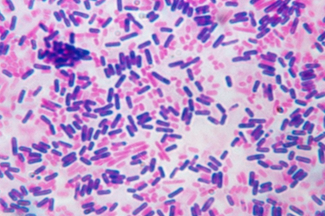
Growth medium is used for support of micro-organisms, cells and small plants. Microbial growth medium can be solid or liquid depending on the type of Micro-organisms to be grown and cultured. It is necessary to select an appropriate culture medium for in-vitro cultivation.
Components and media Formulation:
The requirement of media components depends on the type of cell lines used. Every component used has a specific role to perform. Basic components of media are glucose, amino acids, salts, vitamins, other nutrients, energy source, nitrogen source, water and carbon source.
Carbon and Energy Sources:
- Carbon metabolism has a role to play in product synthesis. Formation of products directly depends on the rate at which carbon is metabolized.
- Carbohydrates, oils and fats and hydrocarbons are some of the common carbon sources.
- Carbohydrates:
- Most commonly used source of carbon in fermentation processes.
- Maize, cereals and potatoes contains starch, which is an essential carbohydrate. Primarily used in fermentation of alcohol.
- Barely grains contains are rich in amount of carbohydrates like sucrose, cellulose, starch and other sugars.
- One major source of sucrose is sugarcane and Molasses. Molasses(obtained after refining of sugarcane or sugar beets) are one of the major source of carbohydrates.
B. Oils and Fats:
- Oils provide more energy compared to sugars. Vegetable oil is a common source of carbon.
- Oils and fats are generally used more as additives rather than as sole carbon source. It also has anti-foaming properties.
- Examples include: vegetable oil, olive oil, linseed oil, soya been oil.
C. Hydrocarbons:
- Hydrocarbons used as carbon sources are C12- C18 alkanes.
- They have more carbon and energy content per weight when compared to sugars. They are relatively cheap.
- Can be used in antibiotics, organic acids, amino acids and protein fermentation.
Nitrogen Sources:
- Most commonly used nitrogen sources includes, Ammonia, ammonium salts and urea.
- For pH control, ammonia is used.
- Soya meal, corn-steep liquor, peanut meal, cotton seed meal, amino acids and proteins.
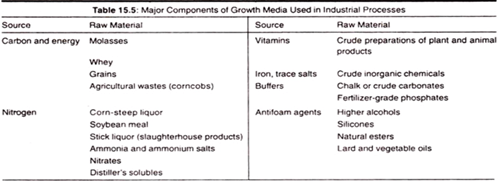
Essential Metals and Minerals:
- Na, K, Ca, P, S, Cl, Mg and Fe are known as macronutrients and are required in larger quantities.
- Zn, Mn, Br, B, Cu, Co, No, V, and Sr are the micronutrients required in relatively less amounts.
- Concentration of these elements used depends on the type of Micro-organisms to be cultured.
Inorganic Salts:
- helps in maintaining osmo-regulatory balance.
- Helps to Increase membrane potential by providing calcium, phosphate and sodium ions.
Growth Factors: common growth factors includes Vitamins, Minerals and fatty acids.
- Amino acids:
- Amino acids being building blocks of proteins, becomes an essential component of any type of culture media.
- Microbial cells cannot synthesise Essential amino acids, and hence it must be supplied for cell proliferation.
- Nitrogen for NAD and NADPH, and nucleotides is provided by L- Glutamine.
- As L-glutamine is an unstable amino acid, it gets converted to other form with time and hence it must be added to medium just before use.
- Non-essential amino acids can also be provided for those that are deprived of growth.
B. Vitamins:
- Cells cannot synthesise vitamins in sufficient quantities and must be supplied. Vitamins serves as important growth factor for cell proliferation.
- B group vitamins like Thiamine, niacin Pantothenic acids are commonly added.
- Serum is the major source of vitamin in media.
C. Fatty acids: mostly important in serum free media, as fatty acids are commonly present in serum. Example- Linoleic acid.
Chelating Agents:
- Insoluble metal precipitation can be prevented by using chelating Agents.
- Chelating Agents forms complexes with metal ions present in media, and than this can be utilised by micro-organisms.
- Example- EDTA( calcium and magnesium complex), citric acid, pyrophosphates.
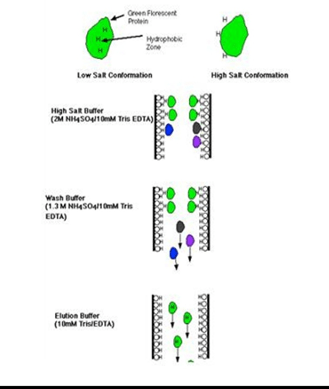
Buffers: role of buffer is to regulate the pH of medium. Micro-organisms growth is affected by changes in pH and hence role of buffers is vital in any type of media Formulation.
- Natural Buffer system– Balance of CO2 along with co3/hco3 content of culture medium is termed as natural Buffer system. Air atmosphere has to be maintained in natural buffering system.(5-10% CO2).
- HEPES: can be used as buffering system. It increases the sensitivity of the media towards phototoxic effects.
- Phenol Red: it is used as an indicator in most of the commercially available media. pH of medium changes due to release of metabolites, as pH changes colour of the solution also changes. Phenol Red turns medium yellow at lower pH, while at higher pH it turns the medium purple.
Anti-foaming Agents:
- Large amounts of foam is produced during microbial processes. Because of excess foaming cells gets removed from the media and leads to Autolysis.
- Hence Anti-foaming agents are required to stop excess foaming and prevent cells from Autolysis.
- Examples- Stearyl alcohol, cotton seed oil, linseed oil, silicones and sulphonates.
Selective agents:
- Mostly antimicrobials are used. This agents makes them selective for certain Micro-organisms.
- They are added in a fixed concentration which is specific and prevents growth of unwanted Micro-organisms.
- Examples- Selenite, bile salts, dye stuffs.
Gelling Agents:
- Most commonly used gelling agent is Agar. It is mostly obtained from sea weeds like Gracilaria and gellidium.
- Gelatine, polyacrylamide, carrageenan scan also be used as gelling Agents.
Serum:
- Most important component of cell culture media. It is a complex mixture of Albumins, growth factors and growth inhibitors.
- For cell cloning and fastidious growth of cell, fetal serum is used.
- Due to its lower growth promoting ability, calf serum, is used for contact inhibition studies.
- 2-10% of serum is present in normal media.
- Serum provides multiple components like proteins(fibronectin), Albumins, amino acids, provides protease inhibitors(protects cells from proteolysis) and it can also act as buffer.
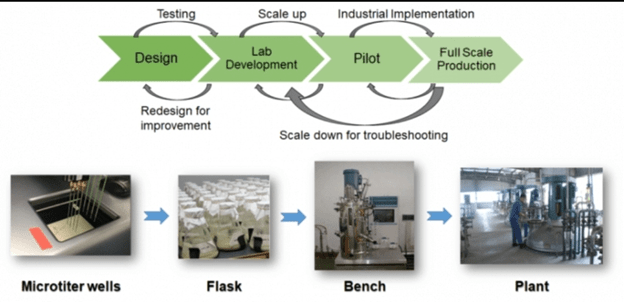
Scale Up of industrial Microbial process:
- According to its name, Scale up simply means increasing something (process) in terms of size, production and it’s amount.
- Scale up of industrial Microbial processes is essential to meet the customer needs and to mass produce a particular product or process for larger profit gains and for greater supply needs.
- For scale up, any process developed in laboratory has to be converted into full manufacturing scale process. For example- 20000- 2000,000L fermenters are used for scale up process.
- Scale-up industrial process is done in 2phases generally:
Pilot Scale- 100-10,000 L fermenters and downstream equipments. The main purpose of pilot Scale- is to convert lab based process to a smaller version of manufacturing process with medium production and scale up.
Demo Scale- 10,000-100,000 L fermenters, with downstream equipments. It minimises the larger investments risk by continuous supply chain, process validation and fulfilling market demand.
Roadmap for proper and successful scale-up process (For large scale plant)
- For an extended period, fully integrated process, including recycle streams can be operated with full industrial materials and equipments.
- Different design models of equipments such as fermenters and suppliers can be evaluated.
- More number of people can be trained to operate large scale plant. Proper operation methods, if followed can lead to faster production.
- Operating-know how and pilot plant data is used to create solutions and planning for preparing large scale plant.
- Large quantities of product can be produced at end application, to build healthy customer relationship and increasing demand for higher commercial plant output.
Scale-up process is essential in terms of increasing production and fulfilling the demand based on increasing needs and supply chain of products.