BY: SHREELAKSHMI
The sudden inheritable changes in the genetic materials are called mutations.
Mutations may be harmful, beneficial or neutral in their effect. Majority of mutations are harmful, because most of the organisms are already adapted and any changes would be disadvantageous. But some mutations are beneficial.

CLASSIFICATION OF MUTATION
1. GENE MUTATION OR POINT MUTATION
These are changes that occur in the fine structure of the genetic material. These changes are also heritable. Usually it involves a single nucleotide or nucleotide pair.
2. CHROMOSOMAL MUTATIONS
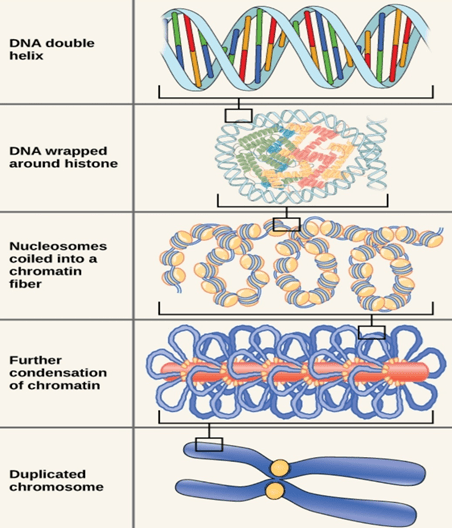
These are changes that affect larger regions of a chromosome and the number of chromosomes. Such changes in the structure and number of chromosomes can be observed under microscope. It is associated with the appearance of new traits in organisms. Five types of chromosomal mutation:
- Deletion-due to breakage apiece of chromosome is lost
- Insertion-chromosomes segment breaks off, flips and reattaches.
- Duplication-Gene sequence is repeated.
- Translocation-Part of one chromosome is relocated with another chromosome.
- Non Disjunction-Gametes will contain too many chromosomes due to the failure of separation during meiosis.

TYPES OF MUTATIONS
- Somatic and Germinal Mutations: If mutation occur in the somatic cells (non-reproductive cells) it is called somatic mutation. If a mutation occurs in the reproductive cells it is called germinal or genetic mutation.
- Dominant and recessive mutation: when a mutation produces a dominant phenotypic expression it is called dominant mutation. If a mutation produces a recessive expression, it is called recessive mutation.
- Micro and macro mutation: When mutation produces small visible changes, e.g., white ye in Drosophila. Such mutations are called micro mutation. Mutations which produce prominent visible changes, e.g., albino maize, Ancon Sheep etc. are called macro mutations.
- Spontaneous and Induced Mutations: Naturally occurring mutations are called spontaneous mutations. Mutations produced artificially by action of certain agents are called mutagens. Such mutations are called induced mutations.
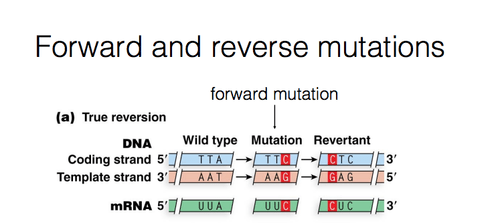
- Forward and reverse mutations: During mutation, a change take place from the normal type (wild type) to a new form (mutant form).This is called Forward mutation. The mutants may revert to the wild forms. Such changes are called reverse or back mutations.
- Lethal mutations: Some mutations affect the vital functions of an organisms. This leads to the death of an organism .Such mutations are called lethal mutations.
MUTAGENS
Mutagens are substances which induce mutation. Such mutations are called Induced mutation. The frequency of such mutations is higher than spontaneous mutations. Mutagens are classified into two major groups:
1. High Energy Radiations
2. Chemicals
Applications of Gene Mutation
G.J .Mendel could not have put froth the basic ideas on inheritance, had all the pea plants were only tall etc.
T.H Morgan was able to evolve his ideas about sex-linked inheritance only when he could find out the mutant white-eyed Drosophila flies.
T.Benzer could analyses the fine structure of genes with the help of mutant forms of T4 phages.
Plants with new and beneficial traits have been evolved through induced mutations. Plant breeders have produced mutants in barley, wheat, oats, soybean, tomatoes etc., which have desired characters like increased yield and resistance to disease.
The yield of Penicillium has been increased through mutation.