Mitosis is a type of cell division that takes place in living organisms and it is commonly defined as the process of duplication of chromosomes in eukaryotic cells and distributed during cell division.
The process where a single cell divides resulting in two identical cells, each resulted cell contains the same number of chromosomes and same genetic composition similar to the parent cell.
Mitosis was first discovered in plant cells by Strasburger in 1875. In 1879, mitosis is also discovered in animal cells by W. Flemming. Flemming in 1882 gave the term Mitosis.
The term mitosis is derived from the Greek word such as ‘Mitos’ means thread.
OCCURRENCE OF MITOSIS:
The mitosis takes place in somatic cells. The cells which undergo mitosis are called Mitocytes.
In plants, the mitocytes are called meristematic cells. Some of the major sites of Mitosis in plants are root apex, shoot apex, intercalary meristem, lateral meristem, leaves, embryo, and seeds.
In animals, the mitocytes are stem cells, germinal epithelium, and embryonic cells. In animals, it mainly takes place in Embryo, skin, and bone marrow.
Mitosis also occurs during the regeneration of the cells. The mitosis takes place for three main reasons such as growth, repair, and asexual reproduction.
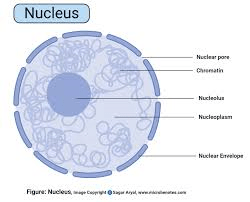
NUCLEUS:
It is a membrane-bound cell organelle present in both animal and plant cells. It is the center of the cell where genetic material is stored in the form of DNA. The DNA is arranged into a group of proteins into thin fibers. During the Interphase of the cell division, the fibers are uncoiled and dispersed into the chromatin. During mitosis, these chromatin condenses to become chromosome.
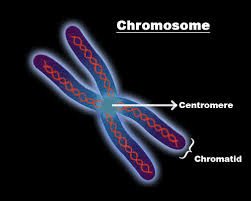
CHROMOSOME:
The chromosomes carry genetic material and they are made of DNA. The mitotic chromosomes possess two sister chromatids, they are narrow at the centromere. They also contain identical copies of original DNA. These Mitotic chromosomes are homologous, they are similar in shape, size, and location of centromere.
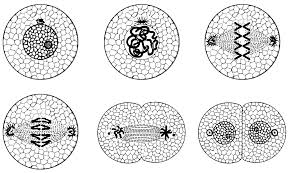
STAGES OF MITOSIS CELL DIVISION:
The mitosis cell division is broadly explained in two stages such
Karyokinesis: Division of Nucleus. Greek ‘karyon’ means the nucleus, whereas ‘kinesis’ means movement.
Cytokinesis: Division of Cytoplasm.
KARYOKINESIS:
4 different stages that take place in Karyokinesis.
1.Prophase
2.Metaphase
3.Anaphase
4.Telophase
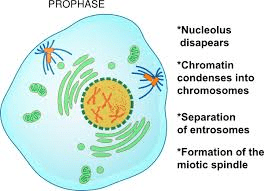
PROPHASE:
The nucleus becomes spherical and the cytoplasm becomes more sticky.
The chromatin slowly condenses into well-defined chromosomes.
During Prophase, the chromosomes appear as a ball of wool. The chromosomes consists of two threads which are longitudinal known as chromatids.
The chromosomes appear as two sister chromatids joined at the centromere.
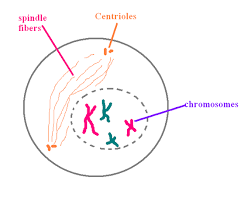
The microtubules are formed outside the nucleus.
In plant cells, the spindle apparatus is formed without centriole. In animal cells, the centriole is divided into two moves towards opposite poles.
METAPHASE:
Nuclear envelop breaks down into membrane vesicles and the chromosomes are set free into the cytoplasm.
Chromosomes are attached to spindle microtubules through kinetochore.
Nucleolus disappears.
Kinetochore microtubules arrange the chromosomes in one plane to form a central equatorial plate.
Centromeres lie on the equatorial plane while the chromosome arms are directed away from the equator called auto orientation.
Smaller chromosomes remain towards the center while larger ones arrange at the periphery.
Metaphase is the longest stage of Mitosis and takes place for about 20 minutes. It is the best stage to study the structure of chromosomes.
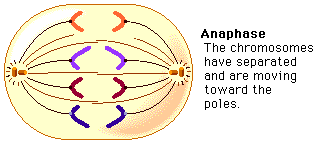
ANAPHASE:
Chromosomes split simultaneously at the centromeres so that the sister chromatids separate.
The separated sister chromatids move towards the opposite poles.
The daughter chromosomes appear in different shapes such as V-shaped(metacentric), L-shaped(sub-metacentric), J-shaped(acrocentric), rod-shaped(telocentric).
The spindle fibers are attached to the centromere and pull the chromosomes to the poles.
Anaphase is the shortest stage of Mitosis.
TELOPHASE:
Daughter chromosomes arrive at the poles. Kinetochore microtubules disappear.
Chromosomes uncoil into chromatin.
Nucleolus reappears. The formation of nuclear envelope occurs around each pair of chromosomes.
The Viscous nature of the cytoplasm decreases.
Telophase is called a reverse stage of the prophase.
CYTOKINESIS:
Cytokinesis is defined as the division of cytoplasm.
It starts during the anaphase and is completed by the end of the telophase.
It takes place in 2 different methods.
a) Cell plate method: It takes place in plant cells. The vesicles of Golgi fuse at the center to form a barrel-shaped phragmoplast. The contents of the phragmoplast solidify to become a cell plate, this cell plate separates the two daughter cells.
b) Cleavage or cell furrowing method: It takes place in Animal cells. In this method, a Cleavage furrow appears in the middle, which gradually deepens and breaks the parent cell into two daughter cells.
SIGNIFICANCE OF MITOSIS:
Mitosis is called as an equational division in which daughter cells produced are identical.
It maintains the constant number of chromosomes and genetic stability in somatic and vegetative cells of the living organisms.
It helps to increase the cell number so that zygote transforms into a multicellular adult.
Healing wounds takes place by Mitosis.
It helps in asexual reproduction.
Mitosis is necessary for growth, maturity and to repair damaged cells.
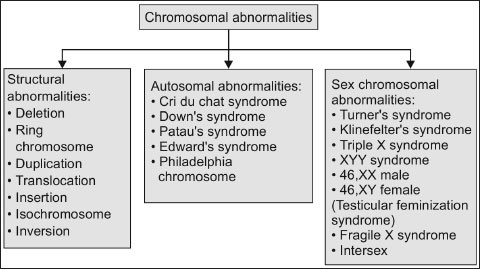
Chromosomal abnormalities are seen in many people. These are generally aneuploidy (presence of abnormal number of cells). it’s either autosomal or sex-chromosome. Translocation, duplication and deficiency in chromosome can also be the reason for this abnormality. It is found that certain disorders or diseases in man are associated with the abnormal number and nature of chromosome.
Abnormalities in Sex Chromosome
A normal male and female has XY and XX chromosome respectively.Klinefelter’s syndrome and Turner’s syndrome are an example for the anomalies in the number of sex chromosomes. Other important abnormal sex in man is the XYY male. They are usually tall and are more aggressive. Many cases it is observed they are mental retarded, but they are fertile.
Abnormalities of the Autosome
In some cases abnormal number of some of the autosomes or other autosomal aberrations. First autosomal anomaly that was described in man is Down’s syndrome. This abnormality is also called mongolism, as the features of such individuals resemble those of Mongolian races. This syndrome was first described by Seguin in 1844.The symptoms include: round face with small skull, short body, swollen tongue, eyelid fold resembling those of Mongolian’s and also mental retardation. In some cases congenital heart diseases is present in these individuals and mostly death occurs early in childhood. But some of them live upto young adult age with the low IQ.
Studies showed that Down’s syndrome is due to trisomy of the 21st chromosome. This trisomy is due to non-disjunction during meiosis. It may occur either during spermatogenesis or oogenesis. Sometimes the third chromosome in the set 21 may be attached to another autosome which is most commonly paired to .This is called translocation trisomy. Down’s syndrome can be related to the age of mother. Frequency of this syndrome increases with increase in mother’s age. In most cases, Down’s syndrome births occur in women conceiving after the age of 30.The trisomy’s of two slightly larger chromosome number 13 and number 18 which leads to early death.
13-Trisomy: It is observed less in number. Such people will have severe mental and physical deformities. The head and the eyes are small.in some cases eyes are absent. Such individuals have congenital heart disease. This disease is also called Patio’s syndrome. This syndrome can be also due to isochromosme of chromosome-13.
18-Trisomy: They show low set ears, simian crease, deformities of fingders, toes and feet, congenital heart disease and receding chain. Head will be flattened 90% of people die within one year.it is also called Edward’s syndrome.
Abnormalities of Chromosome Structure
Chromosomal abnormalities like deletions, translocations, ring formation etc. Are lethal even as heterozygotes, resulting in zygotic rules, still births or infant deaths. Sometimes infants with small chromosome deficiencies survive. However these infants die at an early age.
Cri-du-chat syndrome: It is due to the chromosome deficiency in the short arm of chromosome 5. This deficiency is designated as 5P. Infants with this syndrome cry like a cat mewing. Other features are small forehead, broad face with saddle nose, widely spaced eyes with epicanthic folds and physical and mental retardation.
Chronic Myelocytic Leukemia: This disease is caused when there is a deletion of chromosome 22. Exposure to X-rays can be a cause of this disease.
GENETIC DISEASES DUE TO DEFECTS IN THE CATABOLISM OF PHENYLALANINE
Phenylketonuria (PKU): The conversion of phenylalanine into tyrosine is mediated by the enzyme phenylalanine hydroxylase. The absence of this enzyme causes a disorder in a man.It is called phenylketonuria. In people affected with this disorder,phenyalanine will not be converted tyrosine.Instead,phenyl-acetic acid(PAA) are formed.Phenylpyruvic acid causes damage to nervous system which leads to mental disorder.it is a serious disease and can be detected by simple blood and urine test.Affected infants can be given a diet that is low in phenylalanine.
Alkaptonuria: This disease is caused due to the accumulation of homogentisic acid. Homogentisic acid is responsible for the oxidation of phenylalanine during catabolism. Absence of enzyme responsible for the oxidation of homogentisic acid, it gets accumulated and passes into the urine. When such urine is exposed to air, alcoptone gets oxidized and forms pigments. The absence of this oxidase enzyme is due to to air, alcaptone gets oxidase enzyme is due to and a recessive genes.Alkaptouria is not a serious gene.
Albinism: The color of the skin and the hairs is due to the presence of a dark pigment called melanin. Individuals who do not have melanin are pale in colour.Such individuals are called albinos. This is due to a metabolic block in the conversation of tyrosine into melanin. This block is due to the absence of the enzyme catalyzing the conversion of tyrosine into DOPA (3, 4-dihydroxy phenylalanine).This enzyme is called tyrosine, Albinism may be due to deficiency in the processes that takes place after the formation of DOPA.
Sickle-cell Anaemia
The anaemic condition of the person due to change in the blood cells is called sickle-cell anaemia.It is a chronic, haemolytic disease.In the past, patients died early due to infections or cardiac failure. The sickled red cells become trapped in the small blood vessels. This causes impaired blood circulation resulting in the damage of vital organs. The sickle cells are more fragile and hence haemolyse readily, so have a shorter life than the normal cells. They will suffer from severe anaemia.They are generally genetically transmitted. When two individuals who have sickle – cell trait marry 25% of the progeny will have sickle-cell anaemia, 50% will have sickle –cell trait (carriers) and 25% will be normal individuals.
AMINOCENTESIS
It is prenatal detection of inherited diseases. Human fetus lies in the amniotic cavity in the uterus of the mother. A fluid, amniotic fluid, surrounds the fetus and fills the amniotic cavity. Cells from the fetus are shed into this fluid. The amniotic fluid along with the fetal cells can be collected by introducing a syringe through the abdominal wall of the mother after the third month of pregnancy. This procedure is called amniocentesis. This amniotic fluid is further analyzed.
The fetal cells separate from the amniotic fluid are called amniotic cells. Identification of sex of the fetus helps to terminate pregnancy in people who have a history of sex-linked diseases such as hemophilia, color blindness. Turner’s syndrome and klinefelter’s syndrome can be detected by observing the number of Barr bodies.
Amniotic cells can be cultured in an appropriate medium for 2-6 weeks and can be used for chromosome analysis and biochemical studies. Chromosome analysis helps to determine the karyotype of the foetus .When any kind of an abnormality is found the mother is asked for abortion which is legal.
Disadvantage of Amniocentesis
It can be done only after 12-16 weeks after conception. Analysis of the culture will take another 2-6 weeks.Therefore, if any kind of abnormalities are found and the mother is asked for abortion it will be too late.Aminocentesis will give a mild shock to the fetus affecting its growth.
Apoptosis or Programmed cell death. It is an induced and ordered mechanism in which the cell is actively involved in bringing about its demise. This is a crucial factor in the homeostatic control of many cell populations, including hematopoietic ones.
1. Cells undergoing programmed cell death also show distinctive morphological changes, collectively called apoptosis.
2. These changes include a marked decrease in cell volume, cytoskeleton alteration resulting in membrane blebbing, chromatin condensation, and DNA degradation into smaller fragments.
3. Following these morphological changes, an apoptotic cell sheds tiny apoptotic bodies containing intact organs.
Macrophages rapidly phagocytose apoptotic bodies and cells in advanced apoptosis. This ensures that their intracellular content is not released into the surrounding tissue, including proteolytic and other lytic enzymes, cationic proteins, and oxidizing molecules. Apoptosis causes no local inflammatory response. Apoptosis varies markedly from necrosis, cell death associated with an injury. In necrosis, the wounded cell swells and bursts, releasing its contents and likely causing an inflammatory response.
Each hematopoiesis-produced leukocyte has a characteristic lifetime and then dies by programmed cell death. For example, there are around 5 ×1010 neutrophils in the circulation in adults. These cells only last a few days before programmed cell death is triggered. Together with constant neutrophil development, this death maintains a stable number of these cells. If programmed cell death fails, leukemia can develop. Also, programmed cell death plays a part in holding decent numbers of hematopoietic progenitor cells. For example, in removing colony-stimulating factors, progenitor cells undergo apoptosis. Beyond hematopoiesis, apoptosis is critical in tolerance and killing target cells by cytotoxic T cells or natural killer cells.
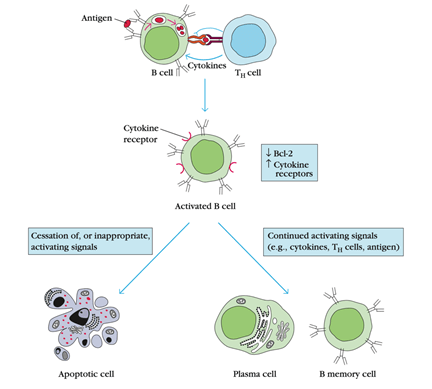
Regulation of Activated B-cell numbers by Apoptosis :
1. Bcl-2 levels were found to play an essential role in controlling the expected lifespan of different hematopoietic cell lines, including lymphocytes.
2. An average adult has around 5 L of blood with about 2000 lymphocytes / mm3 for around 1010 lymphocytes.
3. During acute infection, the lymphocyte count increases 4- to 15-fold, resulting in a total lymphocyte count of 40 ×50 ×109.
4. Since the immune system cannot withstand such a massive increase in cell numbers for a prolonged time, the system needs to remove excess activated lymphocytes after the antigenic threat has passed.
5. Activated lymphocytes express lower levels of Bcl-2 and are thus more vulnerable to apoptotic death induction than naive lymphocytes or memory cells.
6. However, if antigen activates the lymphocytes, then the signals obtained during activation block the apoptotic signal. As antigen levels subside, block activation and lymphocytes begin to die from apoptosis.
Several gene expressions accompany leukocyte apoptosis and other cell types. Some proteins specified by these genes are apoptosis induced; others are essential during apoptosis, while others inhibit apoptosis. For example, radiation can cause apoptosis in thymocytes, but only if the protein p53 is present; Fas signals cause many cell deaths, a molecule found on the surface of several cells, and proteases known as caspases are involved in a cascade of reactions that lead to apoptosis. By contrast, the family members of the bcl-2 (B-cell lymphoma 2) gene, bcl-2, and bcl-XL encode protein products that inhibit apoptosis. Interestingly, in studies involving non-cell death but an uncontrolled proliferation of B cells in the form of cancer called B-lymphoma. bcl-2 was recognized as the first member of this gene family. The bcl-2 gene was at the break-point of chromosomal translocation in human B-cell lymphoma. The translocation transferred the bcl-2 gene into the immunoglobulin heavy-chain locus, resulting in transcriptional activation of the bcl-2 gene and lymphoma cells overproduction of the encoded Bcl-2 protein. The resulting high Bcl-2 levels are thought to help transform lymphoid cells into cancer cells by inhibiting signals that would typically induce apoptotic cell death.
Western blotting, also known as immunoblotting. This method is used for detecting proteins and post-translational protein changes, using antibody-based samples to extract precise protein information from complex samples.
It’s a standard approach in molecular biology, biochemistry, and cell biology with several applications. It can provide semi-quantitative or quantitative protein data in simple or complex biological samples.
Western Blot is also used to separate and identify proteins. In this process, a protein mixture is separated by gel electrophoresis based on molecular weight. These observations are then transferred to a membrane that produces a band for each protein. The membrane is then incubated with protein-specific antibody labels of interest.
The unbound antibody is washed away, leaving the protein of interest with only the bound antibody. By developing the film, the bound antibodies are then detected. Since the antibodies only bind to the protein of interest, there should be only one band visible. The band’s thickness corresponds to the amount of protein present in the sample.
Sample Preparation :
1. Cell lysates are the most common type of sample used in the western blot technique. In the cell cytosol, protein extraction aims to gather all the proteins. This can be achieved with protease inhibitors at a cold temperature to avoid protein denaturing.
2. Using a spectrophotometer, protein concentration is also measured. Using this concentration, the relationship between concentration, density, and volume allows the density of the protein-loaded into each well to be measured.
3. After deciding the necessary sample amount, it is diluted into a glycerol-containing loading buffer so that the samples quickly fall into the gel wells.
4. There is also a monitoring dye (bromophenol blue) in the buffer, allowing the observer to see how far the separation has advanced.
5. After being diluted into a loading buffer, the sample is heated to denature the higher-order structure while maintaining sulfide bridges. High structure denaturing ensures that the negative charge of amino acids is not neutralized, allowing the protein to shift in an electric field.
6. Having positive and negative controls for the sample is also very relevant.
7. A known target protein source, such as a distilled protein or a control lysate, is used for positive control. This helps to validate the protein’s identity and the antibody’s activity.
Gel Electrophoresis :
There are two different kinds of agarose gels used by western Blot: stacking and separating gel.
Stacking gel: The stacking gel is slightly acidic (pH 6.8), which is present at the top position and has a lower concentration of acrylamide, making a porous gel that poorly separates protein, but allows thin, sharply formed bands on the gel.
Separating gel: The separating gel is basic (pH 8.8), also known as resolving gel, which has a higher polyacrylamide content, making the pores of the gel narrower. Therefore, in this gel, the protein is more differentiated by its size, since the smaller proteins migrate more quickly and thus faster than the larger proteins.
The proteins have a negative charge when mounted on the gel since they have been denatured by heating, and when a voltage is applied, they will migrate towards the positive electrode. Using the buffer solution, gels are typically created by pouring them between two glass or plastic plates. Samples are loaded with a marker, and a sample buffer is loaded into the empty wells. The gel is then attached and allowed to run on the power supply. If the voltage is high, it can overheat and distort the bands.
Blotting: After the separation of protein samples, It is then transferred to a membrane.
Nitrocellulose membrane: Two membrane forms exist: nitrocellulose and PVDF. Nitrocellulose is used because of its high protein affinity and retention capacities. It is brittle, however, and does not permit the membrane to be used for reprobation. PVDF membranes provide better mechanical support in this regard and allow reprobation and storage of the Blot. In the PVDF membranes, however, the background is higher; washing is therefore essential.
1. Using an electric field directed perpendicular to the gel surface, the transition is performed, which allows the proteins bands formed to travel out of the gel and onto the membrane.
2. The membrane is embedded into a sandwich between the gel surface and the positive electrode.
3. To cover the gel and blotting membrane, a fiber pad at both ends and a paper towel are used as a sandwich.
Two aspects are essential here:
a. the close contact between the gel and the membrane to ensure a clear image
b. the location between the gel and the positive electrode of the membrane.
For a successful transfer, the membrane has to be placed so that the negatively charged proteins move from the gel to the membrane. This transfer form is called an electrophoretic transfer, which can be achieved in semi-dry or wet conditions. Wet conditions are typically more stable, as the gel is less likely to dry out, and larger proteins are preferred.
Washing, blocking, and antibody incubation :
Blocking is a very significant step in western blotting, as it prevents the non-specific binding of antibodies to the membrane. To minimize the background, blocking is often made with 5 percent BSA or non-fat dried milk diluted in TBST.
Sometimes, non-fat dried milk is preferred as it is readily available and inexpensive. Even milk proteins are not compatible with all detection markers, so the required blocking solution must be selected with caution.
Example- , BSA blocking solutions are preferred to be milk containing casein, a phosphoprotein, and biotin. Incubating the primary antibody with BSA is also a successful idea since it is generally required in higher quantities than the secondary antibody. If the Blot does not provide satisfactory results, placing it in a BSA solution allows the antibody to be reused.
The antibody concentration depends on the sample used. In a wash buffer, such as PBS (Phosphate buffer saline) or TBST (Tris Buffer Saline with Tween 20), the antibody can be diluted. As it minimizes background and removes unbound antibodies, washing is essential. For a very long time, the membrane should not be left to wash, as the signal can also be diminished.
Using the labeled antibody, the membrane is then detected, usually with an enzyme such as horseradish peroxidase (HRP), which is then detected by the signal as it creates corresponding to the target protein’s location. This signal is recorded in a film that is usually made in a dark space.
Quantification :
It is essential to know that data generated with a western blot is usually called semi-quantitative. This is because it offers a relative comparison, though not an absolute quantity calculation, of protein levels.
There are two causes for this; first, there are differences between the samples in separate lanes in the loading and transfer rates that are different on independent blots. Until a more reliable comparison can be made, these variations would need to be standardized. Second, the detection-generated signal is not linear across the sample concentration spectrum. Therefore, because the produced signal is not linear, the concentration model should not be used.
Fig: Sequential stages of the Western blot process.
Every time a cell divides, DNA breaks each of its double strands into two single strands. Each of these single strands functions as a template for a new complementary DNA strand. Each new cell, as a result, has its complete genome. This method is known as the replication of DNA. Replication is regulated by the pairing of template chain bases with incoming deoxynucleoside triphosphates and is driven by DNA polymerase enzymes. It is a complicated process involving an array of enzymes, particularly in eukaryotes.
1. DNA biosynthesis begins in the direction of 5′- to 3′. This makes it difficult for both strands to be simultaneously synthesized by DNA polymerases. It is first necessary to unwind a portion of the double helix, and helicase enzymes mediate this.
2. The leading strand is continuously synthesized, but in short bursts of about 1000 bases, the opposite strand is copied as the lagging strand template becomes available. The resulting short strands are called Okazaki fragments.
3. There are at least three distinct DNA polymerases in bacteria: Pol I, Pol II, and Pol III; Pol III is primarily involved in the elongation of the chain.
4. However, DNA polymerases may not initiate de novo DNA synthesis, but require a short primer with a free 3′-hydroxyl group. This is provided by an RNA polymerase (also called DNA primase) in the lagging strand that can use the DNA template and synthesize about 20 bases in length for a short piece of RNA.
5. Pol III can then take over one of the short RNA fragments previously synthesized. Pol I take over at this stage, using it’s 5′- to 3′-exonuclease activity to digest the RNA and fill the DNA gap until it reaches a continuous DNA stretch.
6. This leaves a gap between the newly synthesized DNA 3′-end and the DNA 5′-end previously synthesized by Pol III.
7. DNA ligase, an enzyme that creates a covalent bond between a 5′-phosphate and a 3′-hydroxyl group, aims to fill the gap.
Transcription:
The mechanism of producing an RNA copy of a gene sequence is transcription. This version, called a molecule of messenger RNA (mRNA), leaves the cell’s nucleus and enters the cytoplasm, guiding the protein synthesis that encodes.
One of the fundamental processes that happen to our genome is transcription. It’s the mechanism by which DNA is converted into RNA. And you may have heard of the Central dogma of Molecular biology in which DNA is transcribed to RNA and translated to protein. Well, the first part of moving from DNA to RNA relates to transcription. And in unique locations, we transcribe DNA to RNA. But there are many other transcribed RNAs, including transfer RNAs and ribosomal RNAs, which do other genomic functions.
Formation Of pre-messenger RNA:
Initiation: There are similarities between the transcription machinery and that of DNA replication. As with DNA replication, before transcription can occur, a partial unwinding of the double helix must happen, and it is the RNA polymerase enzymes that catalyze this process.
Elongation: Unlike DNA replication, only one strand is transcribed. The strand containing the gene is called the sense strand, while the antisense strand is the complementary strand. A copy of the sense strand is the mRNA produced in transcription but transcribed as the antisense strand.
Termination: Ribonucleoside triphosphates (NTPs), with Watson-Crick base pairing (A pairs with U), align along the antisense DNA strand. For the formation of a pre-m-RNA molecule that is complementary to a region of the antisense DNA strand, RNA polymerase binds the ribonucleotides together. Transcription stops when a triplet of bases read as a “stop” signal enters the RNA polymerase enzyme.
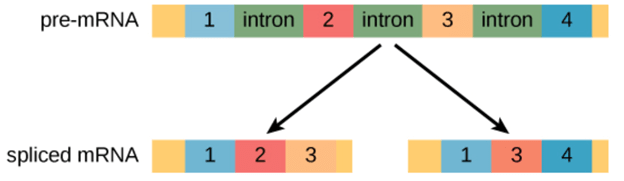
RNA Splicing: Gene expression is regulated at several different stages during transcription and translation to ensure that the right products are produced. The gene includes different sequences in eukaryotes that do not code for protein. The transcription of DNA generates pre-mRNA in these cells. These pre-mRNA transcripts often contain regions known as introns, which are interfering sequences removed by the splicing process before translation. Exons are called the areas of RNA that code for protein. In a process called alternative splicing, splicing can be regulated so that various mRNAs can contain or lack exons. Alternative splicing makes it possible to create more than one protein from a gene. It is a significant regulatory step in deciding which functional proteins are produced from the expression of genes.
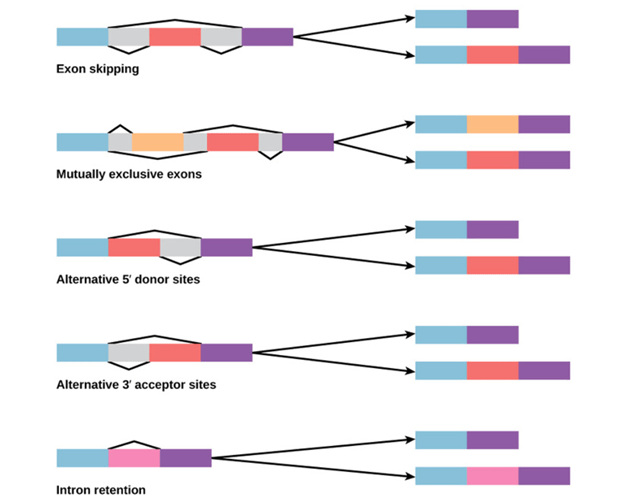
Alternative splicing:
Alternative splicing occurs during gene expression and enables multiple proteins (protein isoforms) generated from a single gene coding process. Due to the distinct forms in which an exon can be exempted from or included in the messenger RNA, alternate splicing can occur. It can also occur if parts of an exon are excluded or included, or if introns are included. For example, if four exons (1,2,3 and 4) are present in a pre-mRNA, they can be spliced and translated into many different combinations. It is possible to translate Exons 1, 2, and 3 together or translate Exons 1, 3, and 4 together.
By binding regulatory proteins (trans-acting proteins that contain the genes) to cis-acting sites located on the pre-RNA, the pattern of splicing and production of alternative-spliced messenger RNA is regulated. Splicing activators (that encourage specific splicing sites) and splicing repressors (that minimize the use of particular sites) are among some of these regulatory proteins.
Heterogeneous Nuclear Ribonucleoprotein (hnRNP) and Polypyrimidine Tract binding protein (PTB) include some common splicing repressors. The proteins translated from; spliced messenger RNAs, alternatively differ in their amino acid sequence, resulting in the protein’s altered function. This is one of the reasons, the human genome can encode a broad range of proteins. A typical process in eukaryotes is alternative splicing; most of the multi-exonic genes in humans are alternatively spliced. Unfortunately, the explanation that there are many hereditary diseases and disorders is often irregular differences in splicing.
Spliceosome:
Messenger RNA splicing is achieved and catalyzed by a macro-molecule complex called the spliceosome. The ligation and cleavage regions are determined by several subunits of the spliceosome, including branch sites and splice sites of 5′ and 3′. Interactions between these sub-units and small nuclear ribonucleoproteins (snRNP) found in spliceosome produce a complex that helps decide which introns to leave out, exons that hold together to bind together. After the introns are cleaved and detached, a phosphodiester bond binds the exons together.
Reverse Transcription: Reverse transcription is a technique used to create a complementary DNA strand from RNA. This mechanism is based on a retroviral mechanism whereby reverse-transcriptase enzymes can reverse transcribe RNA into DNA. This is incredibly helpful when scientists only have tissue and want to study gene sequences. Researchers will separate mRNA from the tissue; in this case, use reverse transcription to generate cDNA.
Translation:
The translation is the process in which proteins are generated using the information carried in mRNA molecules. The nucleotide sequence in the mRNA molecule provides the code for an essential amino acid sequence to create a protein. A protein is made from many amino acids, similar to how RNA is constructed from many nucleotides. A ‘polypeptide chain’ is considered a chain of amino acids, and a polypeptide chain bends and folds on itself to form a protein.
The RNA strand information is translated from RNA’s language into the language of polypeptides during translation, i.e., the nucleotide sequence is translated into an amino acid sequence.
Initiation:
The smaller and larger subunits of the ribosome bind to the mRNA transcript at its binding site during the translation, in the initiation stage.
When the starting codon AUG is recognized by the tRNA, the process of protein construction begins.
Elongation:
The mRNA triplet codon is “read” during the translation elongation process, and the tRNA is added to the complementary amino acid. Ribosomal RNA catalyzes the whole reaction.
Termination:
If the termination codon is reached, the polypeptide chain synthesis ends with peptidyl tRNA. Here, the whole process depends on the RNA polymerase’s involvement in the transcription, although no polymerase is involved in the translation. Interestingly, transcription is a method of encoding information in mRNA (messenger RNA), while translation is a decoding method.
In eukaryotes transcription occurs in the nucleus, while translation in the cytoplasm. However, the entire process occurs only in the cytoplasm in the prokaryotes.
Rifampicin antibiotics inhibit transcription, while translation is hindered by puromycin and anisomycin. The final transcription product consists of Adenine, Guanine, cytosine, and Uracil messenger RNA. At the leading site, it has the initial codon and, in the end, the termination codon. Due to a long adenine chain, the 3 ‘end of the mRNA is called a poly-A tail. The final translation result is a long amino acid chain, a fundamental building block of a protein called the polypeptide chain. The first amino acid is methionine in the amino acid chain. (Although, in most cases, it is removed).
Transfer RNA (tRNA):
1. Ribonucleic acid transfer (tRNA) is a type of RNA molecule that helps decode a sequence of messenger RNA (mRNA) into a protein.
2. During translation, tRNAs act at specific sites in the ribosome, which is a mechanism that synthesizes a protein from an mRNA molecule.
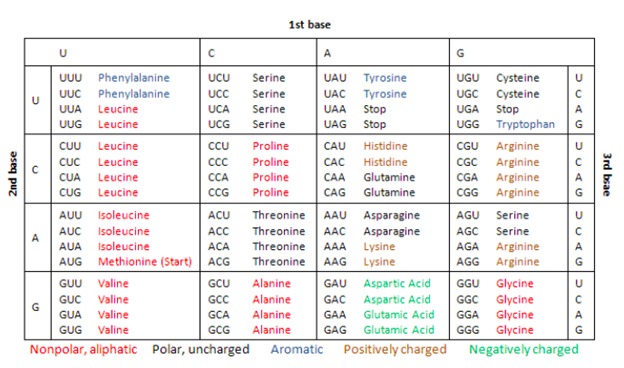
3. Proteins are formed from smaller units called amino acids, which are specified by codons called three-nucleotide mRNA sequences.
4. A codon represents a specific amino acid, and a particular tRNA is known for each codon.
5. With three hairpin loops forming a three-leafed clover, the tRNA molecule has a distinctive folded structure.
6. A series called the anticodon includes one of these hairpin loops, which can identify and decode an mRNA codon. Each tRNA has attached its corresponding amino acid to its end.
7. The tRNA transfers the necessary amino acid to the end of the increased amino acid chain when a tRNA recognizes and binds to its respective codon in the ribosome.
8. Then, the mRNA molecule begins to decipher the tRNAs and ribosomes until the whole sequence is converted into a protein.
Genetic Code: In the early 1960s, American biochemists Marshall W. Nirenberg, Robert W. Holley, and Har Gobind Khorana completed the deciphering of the genetic code. Genetic code is the term we use for the context in which the four DNA bases — A, C, G, and Ts — are linked together to be read and converted into a protein by the cellular machinery, the ribosome. Each of the three nucleotides in a row counts as a triplet in genetic code and codes for a single amino acid. So, each of the three sequences codes for an amino acid. And often, proteins are made up of hundreds of amino acids. Thus, the code that would make one protein could contain hundreds, sometimes even thousands, of triplets.
Blotting is that the technique used for the transferring of nucleic acids or proteins by immobilizing them onto a solid support generally nylon or nitrocellulose membranes. Blotting of macromolecule is that the central technique for hybridization studies. Macromolecule labeling and hybridization on membranes have formed the thought for a selection of experimental techniques involving understanding of phenomenon, organization, etc. Identifying and measuring specific proteins in complex biological mixtures, like blood, have long been important goals in scientific and diagnostic practice. More recently the identification of abnormal genes in genomic DNA has become increasingly important in clinical research and guidance. Blotting techniques are done to identify unique proteins and macromolecule sequences. They have been developed to be highly specific and sensitive and became important tools in both biology and clinical research. Generally principle behind the blotting methods are fairly simple and typically contains four separate steps: electrophoretic separation of protein or of macromolecule fragments within the sample; transfer to and immobilization on paper support; binding of analytical probe to concentrate on molecule on paper; and visualization of bound probe. Molecules during a sample are first separated by electrophoresis then transferred on to an easily handled support medium or membrane. This immobilizes the protein or DNA fragments and they provides a faithful replica of the first separation, they also facilitates subsequent biochemical analysis. After being transferred to the support medium the immobilized protein or macromolecule fragment is localized by the utilization of probes, like antibodies or DNA, that specifically bind to the molecule of interest. Finally, the position of the probe which is suppose bound to the immobilized target molecule is visualized usually by autoradiography.
SOUTHERN BLOTTING TECHNIQUE
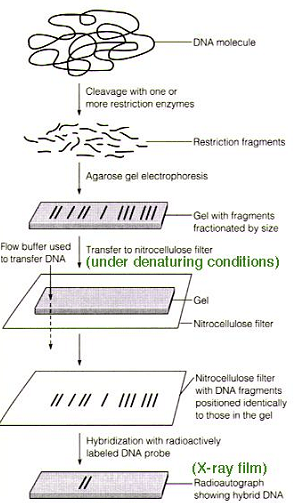
Southern Blot is the analytical technique which is utilized in biology, immunogenetics and other molecular methods to detect or identify DNA of interest from a mixture of DNA sample or a specific base sequence within a strand of DNA.The technique was in 1975 by a biologist E.M.Southern for analyzing the related genes during a DNA fragment and thus named as Southern blotting in his honor.
Principle of southern blotting
The process involves the transfer of electrophoresis-separated DNA fragments to a carrier membrane which is usually nitrocellulose and thus the next detection of the target DNA fragment by probe hybridization. Hybridization refers to the tactic of forming a double-stranded DNA molecule between a single-stranded DNA probe and a single-stranded target DNA. Since the probe and target DNA are complementary to each other, the reaction is restricted which aids within the detection of the precise DNA fragment.
PROCEDURE:
The gene of interest is isolated from the DNA employing a restriction endonuclease .by the action of restriction endonuclease the DNA gets into small fragments during which the specified fragment is additionally present. In order to seek out the proper the fragment agarose gel electrophoresis is performed. Supported the relative molecular mass of strands it get separated during electrophoresis. As we’ve used the double stranded DNA The separated strands also are double stranded it’s converted to single strand the gel is treated with mild alkali like NaOH .It helps in breaking the chemical bond between the double stranded DNA the obtained single stranded DNA is transferred to membrane with the assistance of a transfer solution .IN a tray transfer solution is kept, a glass is kept for the support of gel. On top a nitrocellulose membrane is employed alongside some papers .Above all a weight is kept. Due to the capillarity transfer solution transfers the strands to membrane. To fix the DNA on membrane heat treatment or UV rays are provided. Now the membrane is incubated in a probe solution. Probes are short oligonucleotide sequence used for the detection of molecules. Probes contains radiolabeled molecules which are complementary to the gene of interest. The probes get attached to the DNA strands. This strands are often visualized as using autoradiography to understand whether the gene of interest got transferred to the membrane or not. Southern blotting is additionally called as macromolecule hybridization. As the probe which may be a single strand and therefore the single stranded DNA gets hybridized.
APPLICATIONS:
• Identifying specific DNA during a DNA sample.
• Making of RFLP (Restriction Fragment Length Polymorphism) maps
• Detection of mutations, deletions or gene rearrangements in DNA
• For criminal identification and DNA fingerprinting (VNTR)
• Detection and identification of Trans genes for transgenic individual
• Mapping of restriction sites
• For diagnosis of infectious diseases
• Prognosis of cancer and diagnostic procedure of genetic diseases
• Determination of the relative molecular mass of a fragment and to live relative amounts in several samples.
ELISA stands for Enzyme Linked Immunosorbent Antigen assay. ELISA is an ANTIGEN-ANTIBODY response. In 1971, ELISA was founded. By Peter Perlmann and Eva Engvall at Stockholm university in Sweden. This is a plate-based assay technique used to detect and quantify substances such as peptides, proteins, antibodies and hormones. An antibody conjugated enzyme interacts with a colorless substrate to create a colored substance. This substrate is considered as a chromogenic substrate. A variety of enzymes has been used in ELISA, such as alkaline phosphatase, beta galactosidase and horse-radish peroxidase. Some substrates such as ortho-phenylenediamine dihydrochloride (for peroxidase), p-nitrophenyl phosphate (for alkaline phosphatase), which are hydrolysed by the enzyme referred to above, are used to generate a coloured end product.
In ELISA, an antigen must be immobilised on a solid surface and complicated with an antibody associated with an enzyme. Detection is achieved through an incubation with a substrate to create a measurable product by measuring the conjugated enzymes activity. A highly specific antimicrobial interaction is the most important aspect of the detection strategy.
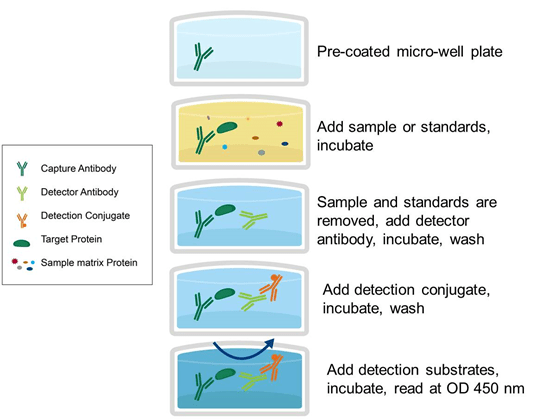
Principle : Usually an ELISA test is performed in a multi-well plate of 96 or 384 well plates. The multi-well plate provides the stable surface for the antigen to stabilise. The immobilisation of the analyte makes it possible to isolate the antigen from the other components in the sample. This function makes ELISA one of the easiest experiments to conduct simultaneously in the multiple samples.
Types of ELISA :
ELISA assays can be found in various formats, based on the binding interactions between antigen-antibody.
They are :
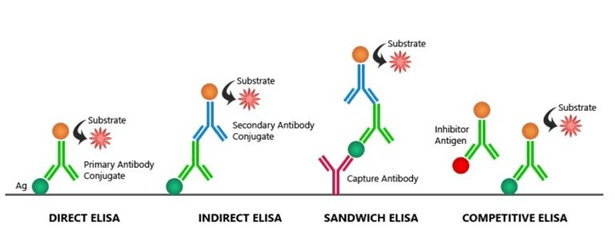
Direct ELISA
Indirect ELISA
Sandwich ELISA
Competitive ELISA
Direct Elisa : the antigen is immobilised and detected on the surface of the multi-well plate with an antigen specific antibody, directly conjugated to HRP or other detection molecules.
Indirect ELISA : With an indirect ELISA, an antibody can be detected or quantitatively determined.
1. An antigen-coated microtiter well is applied to the serum or any other biological sample containing primary antibody (Ab1) and allowed to react with antigen attached to the well. 2. The presence of an antibody bound to the antigen is identified after washing away any free Ab1 by adding an enzyme-conjugated secondary anti-isotype antibody (Ab2), which binds to the primary antibody. 3. Any free Ab2 antibody is then washed away, and an enzyme substrate is added. 4. Specialized spectrophotometric plate readers calculate the amount of colored reaction product that forms and can measure the absorbance of all the wells of a 96-well plate in seconds. 4. This method of choice is for detecting the presence of serum antibodies against Human immunodeficiency virus (HIV), the causative agent of AIDS.
Reaction combinant envelope and core HIV proteins are adsorbed as solid-phase antigens to microtiter wells in this assay. HIV infected individuals can develop serum antibodies to these viral proteins in the epitopes. HIV serum antibodies will usually be detected by indirect ELISA within 6 weeks of infection.
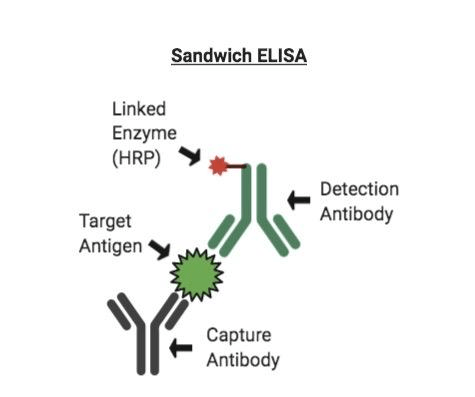
Sandwich ELISA : The antigen can be detected or measured using sandwich ELISA
1. In this procedure, the antibody is immobilised on a microtiter well (rather than the antigen). 2. An antigen containing a sample is added and allowed to react antibody being immobilised. 3. A second enzyme linked antibody specific to different epitopes on the antigen is added after the well is washed and allowed to react with the bound antigen. 4. After washing, substrate is added after any free second antibody is removed and the coloured reaction product is detected.
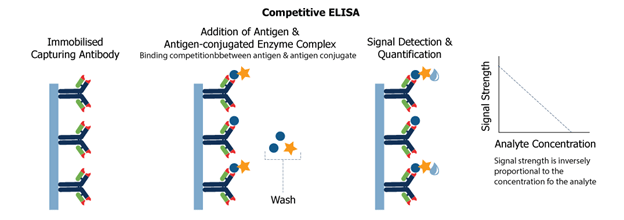
Competitive ELISA : Competitive ELISA is another variation for measuring the amounts of antigen in the sample.
1. In this procedure, antibodies are first incubated in a solution with an antigen-containing sample. 2. To an antigen-coated microtiter well, the antigen-antibody mixture is then added. 3. The more antigen present in the sample, the less free antibody will be available for binding to the well coated with antigen 4. The addition of an enzyme-conjugated secondary antibody (Ab2) unique to the primary antibody isotype can be used to calculate the quantity of primary antibody bound to the well. (as well as in Indirect ELISA). 5. Nevertheless, in the competitive assay, the higher the concentration antigen, the lower the absorbance i.e present in the original sample.
Chemiluminescence : During certain chemical reactions, the measurement of light emitted by the chemiluminescence provides a convenient and highly sensitive alternative to absorption measurements in ELISA that use chemiluminescence. For Example, the oxidation of the luminol compound by H2O2 and the horse-radish peroxidase (HRP) enzyme produces light.
Enhanced sensitivity is the value of chemiluminescence assays over chromogenic ones. In general, by switching from a chromogenic to luxogenic substrate and with the addition of enhancing agents, more than 200 times, the detection limit can be increased at least tenfold. In fact as few as 5moles (5 attomoles) of the target antigen have been detected under ideal conditions.
ELISPOT Assay : ELISPOT Assay is the modification of the ELISA assay. This assay makes it possible to quantitatively determine the number of cells present in a population that contain antigen-specific antibodies or an antigen for which a specific antibody is present.
1. In this method, the plates are coated with the antigen recognised by the antibody of interest or with the antigen-specific antibody which is being tested for production. These specific antigen and antibody are known as capture molecules.
2. The coated plates are then supplemented with a suspension of the cell population under examination and incubated.
3. The cells settled on the plate surface. The secreted molecules that react with the captured molecules, will create a ring of antigen-antibody complexes around each cell that produces molecules of their interest.
4. The plate is then washed, added and allowed to bind an enzyme-linked antibody specific to the secreted antigen or specific to the species ( eg; goat anti-rabbit) of the secreted antibody.
5. Subsequent production of the assay shows the location of the each antigen or antibody producing cell as a point of colour or light by introducing an appropriate chromogenic or chemiluminescence-producing substrate.
Advantages and Disadvantages :
ELISA
Advantages
Disadvantages
Direct ELISA
– Saves time and reagents (Short protocol) – No cross reactivity from secondary antibodies
– In sample all proteins bind to the surface – Primary antibody must be labelled individually. – No signal amplification – Low flexibility
Indirect ELISA
– Has signal amplification – High flexibility
– Long protocol compared to direct ELISA – Potential cross reactivity from secondary antibody.
Sandwich ELISA
– High specificity – Suitable for complex samples. – High flexibility and sensitivity
– Finding two antibodies to the same target that identify different epitopes and function well together even at difficult times.
The recombinant DNA technology has diversified applications in the field of Medicine, Industries, Agriculture, etc.
1.Medicine: In the field of Medicine recombinant DNA technology is one of the important milestones.
a) Production of vaccines: The RDNA technology is employed to produce vaccines by modifying cells and viruses to produce vaccines like the Influenza vaccine, Hepatitis B vaccine, etc. RDNA technique helps scientists to develop vaccines through cloning the gene used for protective antigen protein.
b) Production of human proteins and Hormones: The RDNA technology is used to produce hormones like Insulin, hGH, INF alpha, INF beta, and INF gama, etc. Recombinant DNA technology helped scientists to develop human insulin by using bacteria as a host cell. Now it is even available in the market.
c) Production of HCH: Now a days scientists have developed many growth hormones using rDNA technology, dwarfism is treated with this hormone.
d) Treating infectious diseases: RDNA technology has helped to develop many tests to diagnose diseases like Tuberculosis, Cancer, AIDS, etc. In the diagnosis process, certain pathogens are isolated, identified and then the kits to diagnose are produced when the genome of the specific pathogen is known to kill it or block its pathogenic activity.
e) Production of Interferons: Interferons are virus-induced proteins produced by virus-infected cells. Now, these interferons are produced using rDNA technology. These interferons are used for HCV infection.
f) Diagnosis of diseases: The RDNA technology has provided many tools for doctors to diagnose diseases like food poisoning salmonella, pus-forming staphylococcus, hepatitis virus, and HIV.
2. Agriculture: In the field of Agriculture the rDNA technology plays an important role to improve nutritional value and yield.
Example:
Tomatoes are allowed to ripen on the vine and shelf life of the tomatoes is increased.
BGH hormone produced through rDNA technology allows cattle to gain weight more rapidly and also to produce meat with lower fat content and produce 10% more milk.
Crops sprayed with genetically modified bacteria can tolerate mild freezes.
Organic farmers use Bt toxin to reduce insect damage to crops. This gene for Bt toxin is inserted into various crop plants using rDNA technology.
Gene for beta carotene is inserted into rice crops through rDNA technology.
Production of novel plants with high yielding and pest resistant abilities.
The bacterial genes help in nitrogen fixation can be transferred to cereal crops like wheat, rice, maize, barley, etc.
3. Industry: RDNA technology has numerous industrial applications.
a) Fermentation: The recombinant technology is used to improve strains of microbes which play important role in fermentation.
b) Production of chemical compounds and enzymes which are commercially and industrially important.
c) Developing more efficient strains of microorganisms.
4.Biotechnology:
a) Gene therapy: This RDNA technology helps scientists to replace the defective genes in the genome of the organism. It helps to treat genetic diseases by replacing defective genes in place by normal genes.
b) Creating transgenic animal: Gene of our interest is manipulated and prepared in the laboratory, then transgene is inserted into the selected cattle. Then the transgenic animals are produced.
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR), are the hallmark of a bacterial defines system that forms the basis for CRISPR-Cas9 genome editing technology. In the field of genome engineering, the term “CRISPR” or “CRISPR-Cas9” is often used to refer to the various CRISPR-Cas9 and -CPF1 and other systems that can be edited to target specific stretches of genetic code and to edit DNA at precise locations and also for other purposes, such as for new diagnostic tools. With these systems, researchers can permanently modify genes in living cells and organisms. In the future, it may be possible to correct mutations at precise locations in the human genome in order to treat genetic causes of disease. Other systems now available includes systems such as CRISPR-Cas13 which has the ability to target RNA and provide alternate ways for use and with unique characteristics that have been leveraged for sensitive diagnostic tools, such as SHERLOCK. Francis Mojica who is a microbiologist discovered Crisper.In2017 gene editing experiment on human embryo for the correctness of heart condition was successful. It was done to edit a gene MYBPC3 which caused hypertrophic cardiomyopathy(HCM),which affects 1 in 500.It has no cure and it causes cardiac arrest which leads to sudden death.
HISTORY OF CRISPRs
CRISPRs were first discovered in archaic and later in bacteria’s by Francisco Mojica, a scientist at the University of Alicante in Spain. He stated that CRISPRs serve as part of the bacterial immune system, defending against invading viruses. It consists of repeating sequences of genetic code which was interrupted using spacer sequences which are the remnants of genetic code from past invaders. Mojica’s theory was experimentally demonstrated in 2007 by a team of scientists led by Philippe Horvath. In January 2013, the Zhang lab published the first method to engineer CRISPR to edit the genome in mouse and human cells. Later many of the scientists and teams contributed to the understanding and development of the CRISPR system from the initial discovery to the first demonstrations of CRISPR-mediated genome editing. Feng Zhang and his team have trained thousands of researchers in the use of CRISPR genome editing technology and by sharing more than 40,000 CRISPR components with academic laboratories around the world.
WORKING OF CRISPR
CRISPR mainly uses spacer sequences which are transcribed into short RNA sequences capable of guiding the system to matching sequences of DNA. When the target DNA is found, Cas9 which is one of the enzymes produced by the CRISPR system binds to the DNA and cuts it, shutting the targeted gene off. Modified versions of Cas9 is used by the researchers which can activate gene expression instead of cutting the DNA. These techniques allow researchers to study the gene’s function.
Research also tells that CRISPR-Cas9 can be used to target and modify typos in the three-billion-letter sequence of the human genome which can be helpful in treating genetic disease.
IMPORTANCE OF CRISPR-Cas9 COMPARED TO OTHER GENOME EDITING TOOLS
CRISPR-Cas9 is proved to be an and efficient and tailored alternative to other existing genome editing tools. CRISPR-Cas9 system itself is capable of cutting DNA strands. They do not need to be paired with separate cleaving enzymes as other genome editing tools need. They can also be easily matched with tailored “guide” RNA sequences designed to lead them to their DNA targets. They are generally called as gRNA. Thousands of such gRNA sequences are already created and are available to the research community for studies. CRISPR-Cas9 can also be used to target multiple genes simultaneously, which is another advantage while comparing with other gene-editing tools that.
DIFFERENCE BETWEEN CRISPR-Cpf1 AND CRISPR-Cas9
CRISPR-Cpf1 differs drastically from CRISPR-Cas9 while looking through the angle of research.Cas9 which is a DNA cutting enzyme in its natural form forms a complex which consist of two RNAs which are required for cutting activity. The Cpf1 is a simple system that requires only a single RNA. The Cpf1 enzyme is a smaller than the standard SpCas9 which makes it easy to deliver into cells and tissues.Cpf1 cuts the DNA in different manner than Cas9. The Cas9 complex cuts the DNA into both strands at the same place by leaving the blunt ends that often undergoes mutations while they are rejoining. Whereas Cpf1 complex cuts in the two strands which are offset and leaves short overhangs near the exposed ends. This is done to get help with precise insertion which allows the researchers to integrate a piece of DNA more efficiently and accurately.Cpf1 cuts far away from the recognition site which indicates that even if the targeted gene becomes mutated at the cut site it can still be cut again which allows multiple opportunities for correct editing to be done. The Cpf1 system provides flexibility in choosing the target sites. Similar to Cas9, Cpf1 complex must first get attached to a short sequence known as a PAM, and targets must be chosen which are adjacent to naturally occurring PAM sequences. The Cpf1 complex recognizes very different PAM sequences than those of Cas9. This could be an advantage while targeting, for example the malaria parasite genome and even the human genome.
Scientific uses of CRISPR
CRISPR genome editing allows the scientists to quickly create cell and animal models, which are used by researchers to accelerate their research into diseases such as cancer and mental illness. In addition, CRISPR is now being developed as a rapid diagnostic method.
Among all the advanced techniques in the field of biotechnology gene therapy is one of the important milestones. Gene therapy is a technique used in biotechnology in which new foreign genes are inserted into existing cells of living organisms to treat diseases. This technique is employed to replace the defective genes in the genome which causes diseases. The research on gene therapy was first started in 1972 by Friedmann and Roblin who published a paper entitled ‘gene therapy to cure genetic disorders. Later in the future, the research was developed by scientists, and on the 14th of September 1990, for the first time the gene therapy clinical trials were approved by the National Institute of Health (NIH), the US under the guidance of William French Anderson, which was performed on 4 years old girl who was suffering from severe combined immunodeficiency.
TYPES OF GENE THERAPY:
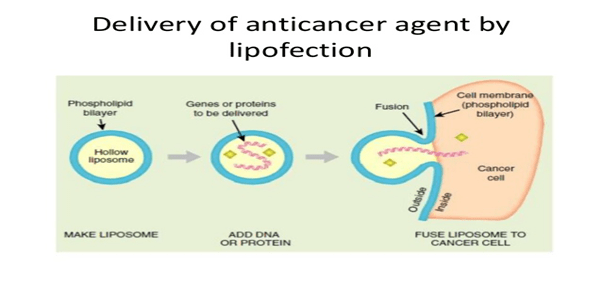
The technique gene therapy is classified into two types based on cells in which foreign gene is inserted, a) Germline gene therapy
b) Somatic cell gene therapy
Germline gene therapy: In this type, the foreign gene or therapeutic gene is inserted into the germ cells of the living organism. In this technique the inserted genes are passed from generation to generation, hence they are heritable.
Example: The therapeutic cells are inserted into the germ cells like sperm and egg.
Somatic cell gene therapy: In this technique, the foreign gene or therapeutic gene is inserted into the somatic cells of the living organism. In this technique, the inserted genes do not pass from generation to generation.
Example: Insertion of genes into bone marrow, blood cells, skin cells, etc.
APPROACHES OF GENE THERAPY:
There are two ways of approaches are available in gene therapy:
a) In-vivo gene therapy
b) Ex-vivo gene therapy
In-vivo gene therapy: In this technique, the target cell is fixed in the body of the living organism and the therapeutic gene or foreign gene is inserted into the target cell through a vector. The vector used to insert the therapeutic gene into the target cell is either a viral or non-viral vector. This technique is mainly used to treat genetic disorders.
Example: It is used to treat the genetic disorder cystic fibrosis.
Ex-vivo gene therapy: In this technique, the therapeutic genes are inserted into the cultured cells in the laboratory.
Procedure: The genetically defective cells are isolated from the living organism and they are cultured inside the laboratory. Then the therapeutic genes are inserted into the cultured cells in the laboratory. After insertion, the genetically corrected cells from the cultured cells are isolated and grown in the laboratory, then the modified cells are transplanted into the living organism.
Example: The first gene therapy was performed on 4 years old girl Ashanthi Disilva, who was suffering from severe combined immunodeficiency (SCID), which is caused due to defects in the gene which codes for adenosine deaminase. This is an example of ex-vivo gene therapy.
VECTORS IN GENE THERAPY:
Vector is the vehicle of gene delivery, an agent that carries to the therapeutic or desired gene into the target cell. There are two categories of vectors are employed in gene therapy.
Viral vectors
Non-viral vectors
Viral vectors: The viruses are used as vectors to deliver the desired gene to the target cell of the living host.
Example: Retrovirus: The genetic material of the virus RNA and it can carry a DNA of size less than 3.4kb and integrated it into the host genome of stable fashion, and it targets the cells in dividing the state.
Adenovirus: The genetic material of this virus is double-stranded DNA, they carry and integrate the desired DNA into target cells that are a non-dividing state.
Non-viral vectors: Some of the chemicals and physical instruments are used to carry desired DNA into the host cell are called Non-viral vectors.
Some of the physical methods to insert the desired DNA into the target cell are Electroporation, gene gun, etc.
Electroporation: In this technique, the electric shock of small pulses of high voltage is used to carry the DNA across the cell membrane of the target cell. After the small electric shock, the temporary pores are caused in the cell membrane to provide the passage for desired DNA into the host cell.
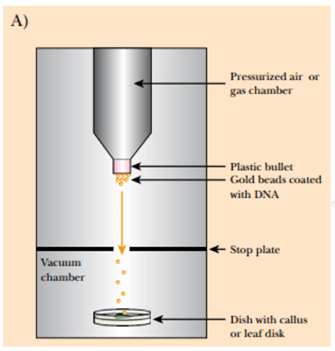
Gene gun: In this method, the gold particles or tungsten particles are coated with the desired DNA molecule, then the coated DNA is placed into a device called Gene gun which generates the high pressure necessary for the penetration of the desired DNA into the target or host cell and the gold and tungsten particles are left behind on stopping disc.
Sonoporation: In this method, the DNA is inserted into the target cell with help of ultrasonic frequencies.
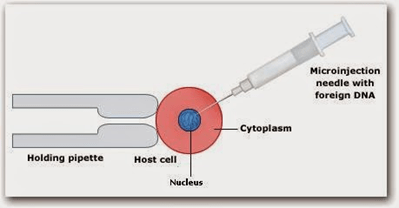
Microinjection: In this method, microscopic materials are inserted into the target cell with the help of a small microscopic device called Micromanipulator.
Some of the chemical methods used to carry desired into the target cell are lipofection and using detergent mixtures.
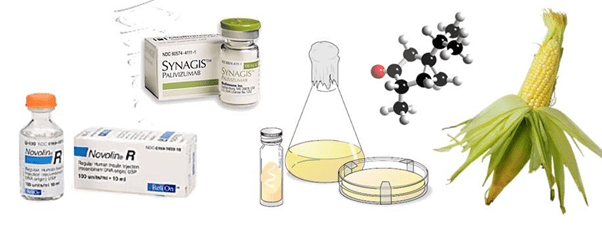
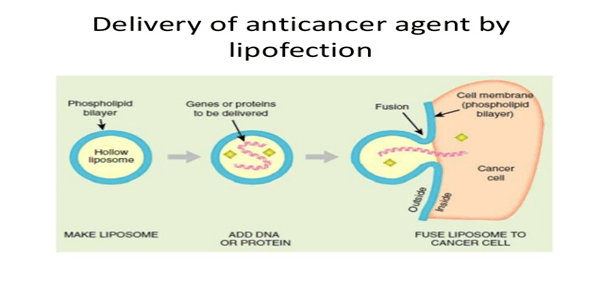
Lipofection: In this technique, the liposomes are used as a vector to insert the desired DNA into the target cell. The liposomes are artificial phospholipid vesicles used to transport various molecules into cells including DNA.
Detergent mixtures: Some of the detergent mixtures like calcium phosphate are mixed with cDNA molecules and they are inserted near to the target cells. Then these detergent mixtures disturb the cell membrane od the cell and increase the size of the pore through which the cDNA enters into the cell.
ADVANTAGES OF GENE THERAPY:
Gene therapy plays an important role to replace the defective disease-causing genes in the genome.
It also helps treat genetic disorders and some of the deadly diseases like cancer.
DISADVANTAGES:
Diseases caused by defects in multiple genes are not treated by gene therapy.
Some of the viral vectors cause toxicity and inflammatory reactions.