Molecular biology is the study of the chemical and physical structure of biological macromolecules. Genetics is a branch of science dealing with the study of heredity and variation.
Molecular Biology is an overlapping with other areas of biology and chemistry. It is the understanding of the interactions between DNA, RNA and protein. It is basically of two steps: transcription and translation. It is called Central Dogma. Synthesis of RNA from DNA is called transcription and synthesis of Protein and DNA translation.
TRANSCRIPTION
Transcription involves synthesis of new strand of nucleic acid complementary to a DNA template strand.
To transcribe a gene, RNA polymerase proceeds through a series of well-defined steps which are grouped into three phases- initiation, elongation and termination.
The bacterial core RNA polymerase can initiate transcription at any point on a DNA molecule.
RNA polymerase can initiate a new RNA chain on a DNA template and therefore do not require a primer.
The elongating polymerase is a processive machine that synthesize and proofreads RNA.
Ribonucleotides enters the active site and the added to are growing RNA chain.
Termination of transcription is activated by the presence of terminator sequence which results in the elongating polymerase to dissociate from the DNA and release the RNA chain.
TRANSCRIPTION
In eukaryotes, they have three different polymerases and several initiation factors.
TRANSLATION
Translation converts the genetic information present within the mRNA to a linear sequence of amino acids in proteins
The decoding of mRNAs into the language of proteins is composed of four components which are Trna,aminoacyl tRNA synthetasesand also ribosome.mRNa template provides the information that must be interpreted.Aminoacyl Trna synthetase couple amino acids to specific tRNAs that recognize the appropriate codon. The protein coding region of each mRNA have contiguous and non-overlapping string of codon called an open reading frame. Translation starts at 5’end and ends at 3’end.it starts with start codon and ends with a stop codon.AUG is usually a start codon, whereas UAG,UGA,UAA are stop codon.
GENETICS
Genetics is the study of hereditary and variation. The term was first introduced by W Bateson.
Gregor Mendel is known as the Father of Genetics.
Mendel was the first one who told that there are some factors which give you a particular phenotype (eye color, hair texture).The pioneering study on generics was by Mendel on pea plants. He looked at how the size, height, colur.He selected only pure breeds. From result of this experiment he came up with the hypothesis:
LAW OF SEGREGATION:
It is studied with the help of monohybrid cross. It states that the alleles of a given locus segregate into separate gametes. It is also called as law of purity.
MONOHYBRID CROSS
LAW OF INDEPENDENT ASSORTMENT:
It can be explained with the help of dihydrid cross. Mendel considered seed form and cotyledon color for the cross. This law states that the factors or alleles of each character assort or segregate independent of the factors of other character at the time of gamete formation and get randomly rearranged in the offspring.
DIHYBRID CROSS
POST MENDELIAN
Basics of genetics was given by Mendel, later studies provided information on various genetic interactions which also led to the studies on various genetic disorders.
COMPLEMENTARY GENES
If two genes present on different loci produce the same effect when present alone but interact to form a new trait when present together .They are called complimentary genes.
SUPPLEMENTARY GENES
They are pair of non-allelic genes one of which produces its effect independently in the dominant state.
LINKAGE
It is an exception of principle of independent assortment.
CHROMOSOME THEORY OF LINKAGE
It was given by Morgan and Castle. It states that
The genes which show linkage are situated in the same chromosome and remain bounded by chromosomes by material.So,they cannot be separated during the processes
The degree or strengthen of linkage depends upon the distance between the linked genes on the chromosomes, closely located genes show strong linkage.
Genes lie in linear order in the chromosome.
CROSSING OVER
It is one of the two exceptions of Mendel’s law of independent assortment. It produces new combinations or recombination of genes by interchanging of corresponding segments between non sisters chromatids of homologous chromosomes at prophase 1 of meiosis the non-sister chromatids in which exchange of segments has occurred are called cross overs.
The plasmids are defined as naturally occurring, stable extra-chromosomal genetic elements found in all three major groups of microbes. (Archaea, bacteria and eukaryotes).
Among eukaryotes, plasmids are found in fungal cells and mitochondria of some plants.
In addition to naturally occurring plasmids, a wide range of plasmids have been engineered for specific applications in Recombinant DNA technologies and in genetic engineering.
Plasmids may be composed of single stranded or double stranded DNA or RNA.
Plasmids may be linear or circular. The size of plasmids range from 1kb to over 200kb. Plasmids replicate independently of the chromosome of cell.
Plasmids provide bacteria with genetic advantages, such as Antibiotic resistance, which is conferred by genes present in the plasmids.
The other important known function of plasmids is they contain genes that enhance survival of organism, either by killing the organism or by producing toxins against it.
Certain plasmids provide selective advantage to the host under specific conditions.
History:
The term plasmid was coined for the first time in the year 1952 by Lederberg.
He worked on experiments with bacterial conjugation to map the genome and revealed two groups of linked genes; (a) Main bacterial chromosomes and (b) a second chromosome that is present only in some bacterial cells.
This class of extra chromosomal elements is termed as Plasmids.
Plasmids are believed to be the evolutionary ancestors of viruses.
Structure of Plasmids:
Plasmids are generally composed of circular double chains of DNA. The two ends of plasmids are held together by covalent bonds.
Origin of Replication (ori): it refers to the site at which replication begins. In plasmids, this ori is generally composed of A-T base pairs, which are much easier to separate during replication. As plasmids are smaller in size, they have one to few origins of replication sites. Regulatory elements are also present at the ori site. For example- Rep proteins.
Multiple cloning sites:this is also called as polylinker. A short DNA sequence consists of few sites for cleavage by restriction enzymes. At the cleavage site, strand can be cut by different polylinkers. One main advantage of multiple cloning sites in plasmids is that it does not hinder the rest of the plasmid during the process and also possess unique restriction enzymes, which can cut the plasmid at specific points to allow DNA insertion.
Antibiotic Resistance gene: This is one of the main components in plasmids which help in Drug resistance. By a process of conjugation, plasmids transfer from one bacteria to the other and during this process they are capable of conferring antibiotic resistance properties to the bacteria.
A Promoter region: this region helps in the process of transcription and in recruitment of transcriptional machinery.
Primer binding site: this is specifically used for PCR amplification or for DNA sequencing and generally refers to short sequence of DNA on a single strand.
Types of Plasmids:
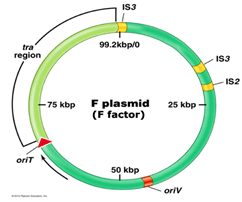
F Plasmid (fertility factor):
It is the best studied conjugative plasmid. It plays a major role in E.coli conjugation.
It is about 100kb in size.
It has genes responsible for cell attachment and plasmid transfer.
Tra operon (has tra genes responsible for nonsexual transfer of genetic material in bacteria) is important in F plasmid.
It contains 28 genes and these genes direct the formation of sex pilus (helps in transfer of DNA in bacteria during bacteria conjugation).
F factor also possesses insertion sequence (short sequences which can acts as transposable elements) that assists plasmid interaction into host chromosome.
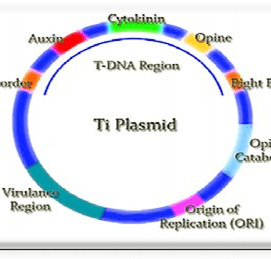
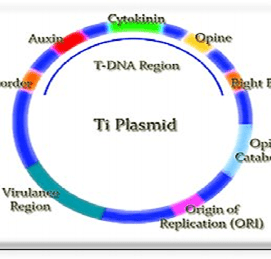
Ti Plasmids:
It is present in soil bacterium, Agro bacterium Tumifaciens.
It induces tumor in plants and hence commonly called as Tumor inducing plasmid.
It is a large plasmid. Generally, its size varies from 180-250kb.
It contains T-DNA region of about 23-25kb. This region is generally transferred into plant cells.
Ti plasmids can be of 3 types based on kind of opines (carbon compounds found primarily in crown gall tumors) they encode for; Octopine, Nopaline, Agropine.
Opines are neither naturally found in plants and nor required by plants. Agro bacterium uses it as a source of carbon and nitrogen for its growth and multiplication.
Mechanism of infection:
Formation of wound is essential in plants for the infection by Agro bacterium Tumifaciens.
The lipopolysaccharides present on the bacterial cell walls and Polygalacturonic acid of damaged plant cell wall helps in the process of attachment of agro bacterium to plant cell.
Damaged plant cell wall also produces Acetosyringone, a low molecular weight phenolic compound. This compound induces transcription of Virulence genes (Vir genes) present on Ti plasmid.
Enzymes produced by virulence genes, makes a nick in one strand of plasmid at two points.
This produces single stranded DNA fragment which is then carried to plant cells.
T-DNA of Ti plasmid integrates with plant cell chromosome. As a result of this plant cells produces opines. This opines helps in growth and multiplication of Ti plasmid.
T-DNA also codes for phytohormones like auxins and cytokinin. This hormones leads to disorganized proliferation plant cells causing tumors called Crown Gall tumors.
R plasmid (resistance plasmids):
R plasmids are well studied group of plasmids. Their role is to confer antibiotic resistance and inhibits various other growth inhibitors.
R plasmids have genes that encode for enzymes that are able to destroy or modify antibiotics.
R plasmids evolve rapidly and can easily acquire additional resistant determining genes.
A single plasmid transfer can turn a drug sensitive bacterium into a multiple drug resistant strain.
Broad host range plasmids that carry multiple antibiotic resistant genes are of great medical concern because they can be transferred to a wide range of bacterial species.
Degradative plasmids:
Degradative plasmids are Plasmids that encode genes required for the metabolism of wide range environmental contaminants.
As they can be transferred between microorganisms, they can provide a means for the rapid horizontal spread of degradative genes among natural microbial populations.
Direct seeding of plasmids by Soil bioremediation by borne genes into native soil is a potential useful way to enhance the degradation of environmental pollutants.
2-4-D plasmids were found in strains isolated by enrichment on 2-4-D as the sole source of carbon and energy and some of them were found to degrade herbicide with similar structure.
Strain of Pseudomonas Putida called NCIB was formed to possess plasmid PDTG1 with 83,042 base pairs. This plasmid also encodes enzymes for Naphthalene degradation.
COL plasmids (col-colicine bacteriocines):
Col plasmids are present in different genes of E.coli.
They contain genes that control the synthesis of proteins called Colicines (proteins which has the ability to kill other bacterial strains and are often used by host bacterium).
This colicines inhibit growth of related bacteria that lacks Col plasmid.
Different types of colicines exhibit different mode of action.
Col-B induces damage of cytoplasmic membrane of the target bacteria.
Example of Col plasmids- (Col E2 and Col E3) causes degradation of nucleic acids.
Col plasmids are may be self transmissible or non-self transmissible (this non self transmissible may be mobilized by F plasmids).
This means that when F+ cell contains Col E plasmid, this plasmid can integrate with F factor and gets transported to F– cell during conjugation.
DNA in a cell is target for various endogenous and exogenous agents that can damage the base or sugar phosphate backbone.
It is estimated that each day 104 or 106 lesions are produced in the DNA of a human cell. Hence, this lesions needs to be repaired in order to avoid mutations in the DNA.
Prokaryotic and Eukaryotic cells have specialized mechanisms to identify and correct various kinds of damage. The rate of this repair depends on the factors such as cell type, age and extracellular environment.
A cell that is no longer able to repair its DNA damage can enter one of these possible states: senescence (old age), Apoptosis (cell death), and Neoplasia (unregulated cell division).
Types of Damages:
There are agents like Endogenous and exogenous agents which can damage the DNA.
There are two major sources of endogenous DNA damage:
Reaction of components with DNA with extremely reactive metabolites, such as reactive oxygen species (ROS) and reactive nitrogen species (RNS) produced from biochemical pathway.
Errors in DNA replication or repair by DNA polymerase.
The main types of damage due to endogenous cellular processes are; single stranded and double stranded breaks, hydrolysis of glycosidic bonds leading to depurination or depyremidation.
Oxidation of bases due to ROS resulting in products such as 8-oxo-G also occurs due to endogenous cellular processes.
Exogenous agents that can cause damage includes:
Cosmic ionizing radiations.
UV solar radiations.
Fungal and plant toxins.
Man made mutagenic chemicals like intercalating agents.
Types of damages generally caused by exogenous agents include; single and double stranded breaks, pyrimidine dimmers, modified bases like ethenobase, oxidized bases.
Inter and intra strand crosslink’s are also caused by exogenous agents.
DNA repair systems and their mechanisms:
Direct Reversal of Damage:
Cells can eliminate three types of base damage by chemically reversing it. This type of repair does not require a template on the complementary strand (normal, non-damaged base).
Photo reactivation by Photolyase:
The absorption of UV light by DNA results in the formation of pyrimidine dimmers, mostly thymine dimmers.
An enzyme called photolyase is activated by energy absorbed from UV light and directly reverses this damage to restore the pyrimidines to their original un-dimerized form.
Photolyase has two types of chromospheres; FADH in all organisms, Folate- in E.coli and yeast. Photolyase is absent in placental mammals.
Mechanism:
The photo antenna in photolyase, N5, N10-Ethenyl-tetrahydro-folyl-polyglutamate absorbs a blue light photon at 300-500nm of wavelength.
It transfers the excitation energy to FADH in the active site of the enzyme.
The excited flavin (FADH) donates an electron to the pyrimidine dimmer. This creates an unstable dimmer radical.
The unstable radical undergoes electronic rearrangement to revert to monomeric pyrimidines.
The electrons are then transferred back to FAD+ to generate FADH.
Methyl Guanine Methyl Transferase:
O6 methyl guanine is a common lesion produced by alkylating agents. It is highly mutagenic because methyl-G tends to pair with with T rather than C during replication. These mispairing results in G: C to A: T transition.
Although alkylating mutagens preferentially modify the guanine base at N7 position, O6 is a major carcinogenic lesion in DNA.
The DNA adduct formed, is removed by the repair protein O6 methyl guanine methyl transferase through SN2 mechanism. Since it removes the alkyl group from the lesion, this protein is not a true enzyme.
The methyl acceptor in the protein is a Cysteine.
Base Excision Repair: (BER)
In this mechanism, damage is caused to a single nucleotide by oxidation, alkylation, hydrolysis or deamination.
There are several specific enzymes in BER mechanism, each able to identify a specific type of base lesion.
These are the DNA N-glycosylases, which can cleave the glycosidic bond between a damaged base and the deoxyribose moiety.
When glycosylase comes across a damaged base, it hydrolyses the glycosidic bond to generate AP (apurinic/apyrimidinic) sites.
Now, the AP endonucleases cleaves the phosphodiester bond at 5’ of AP site, leaving 3’ OH group and a 5’ deoxyribose phosphate residue, marking the abasic site.
This creates a site for binding of DNA polymerase I, which then uses the 3’OH terminus to replace a few nucleotides using the complementary strand as template.
If only one nucleotide is repaired, it is called as short patch repair. If several nucleotides (2-10) are replaced it is called long patch repair.
Most of the BER is via short patch repair.
As the DNA polymerase adds new nucleotides to the 3’end created by AP endonuclease, it displaces the nucleotides 3’ of the nick, leading to Flap.
This flap is cleaved by deoxyribose phosphatase in short patch repair and by flap endonuclease in long patch repair.
Finally, the DNA Ligase seals the nick between the patch filled in by DNA polymerase I and the residue exposed by removal of overhanging.
Nucleotide Excision Repair:(NER)
Nucleotide excision repair- repairs damage affecting longer stretch of DNA, comprising 2-30 bases. The enzymes in NER can recognize bulky, helix-distorting lesions, intra-strand cross links, oxidative damage as well as single strand breaks.
NER has two sub pathways; Global genomic NER and Transcription coupledNER.
NER is critical to survival of all free living organisms; especially those that lack the photolyase system for direct repair of UV induced pyrimidine dimmers.
In E.Coli the key enzyme complex for NER is the ABC exonucease. This complex comprises three subunits, UVrA, UVrB, and UVrC. The complex is called exonuclease as it catalyses two specific endonucleolytic cleavages, one on either side of lesion.
This lesion is detected by a complex of UVrA and UVrB proteins which scans the DNA and binds to the site of the lesion.
Once the lesion is recognized, the UVrA dissociates, leaving behind a tight UVrB-DNA complex.
Now, UVrC binds to UVrB and mediates two incisions.
UVrC mediates the incision at 8th phosphodiester bond on the 5’ side.
UVrB cuts at the 5th phosphodiester bond on the 3’end of the lesion.
Thus, a 12-13 nucleotide long fragment encompassing the lesion is cut out of the DNA.
In the next step, the UVrD helicase removes the 12-13 nucleotide fragment by unwinding it away from the dsDNA.
DNA Ligase seals the nick and complete the process.
Defects in genes for NER proteins results in genetic disorders like, Xeroderma Pigmentosum, Cokaynes syndrome.
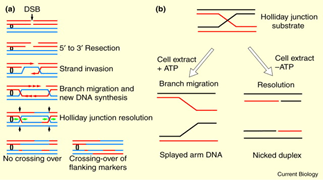
Homologous Recombination Repair:
Double strand breaks of DNA double helix can be repaired by homologous recombination repair.
A DNA cleaving enzyme degrades the broken DNA molecule to generate 3’ end.
The single strand tails thus, generated will invade the unbroken homologous DNA duplex.
Invading strand base pairs with its complementary strand present in the other DNA molecule.
The invading strands with 3’oH group will serve as primers for new DNA synthesis.
Eventually, the second strand also invades and repairs the DNA from 3’end.
This will generate two junctions known as Holiday Junctions. These holiday junctions are produced because of Branch migration.
This recombination intermediate will be further resolved by cleavage.
Mismatch Repair:
This repair mechanism recognizes and repairs errors in insertions, deletions, and in mis-incorporation of bases. It plays a vital role in Homeostasis and genomic stability.
The mismatch is recognized and the DNA is kinked towards major groove. MUTs detects the mismatch with the help of conserved motif, Phe-X-Glu.
MUTs and MUTL interaction occurs and forms a bridge for other protein complexes.
Ternary complex is formed by MUTs, MUTL and mismatched DNA.
MUTS alpha activates EXO1 and removes mismatch base.
DNA repair mechanisms are essential to ensure species survival by enabling faithful inheritance of parental DNA. DNA repair mechanisms if failed to work can lead to cancer and mutations, which may lead to genetic disorders.
In higher eukaryotes, gene expression is tissue-specific. Moderate to high expression of a single gene or only certain cell types show a group of genes. This can be explained through an example, (genes encoding globin proteins are expressed only in erythrocyte precursor cells, called reticulocytes).
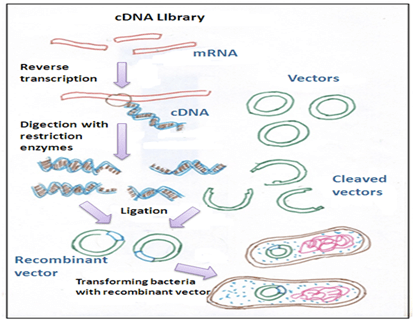
A target gene can be cloned by isolating the mRNA from a specific tissue using this information. Bacteriophage vectors are used to clone specific DNA sequences synthesised from mRNA copies of particular cell type. A fully transcribed mRNA results in cDNA (complementary DNA) which contains only the expressed genes of an organism and replicas (Clones) of such DNA copies of mRNAs are called cDNA clones.
A combination of cloned cDNA fragments constituting some portion of the transcriptome of an organism is termed as cDNA library, which can be inserted into a number of host cells. Before translation into protein, mRNA is spliced in eukaryotic cells. The DNA synthesized from the spliced mRNA does not have introns or non-coding regions of the gene, so because of this, the protein under expression can be sequenced from the DNA.
Following steps are involved in the construction of cDNA library:
1. mRNA isolation
2. First and second strand of cDNA synthesis
3. Incorporation of cDNA into a suitable vector
4.Cloning of cDNAs- the cDNA thus obtained is cloned.
Isolation of mRNA:
Total mRNA from a cell type or tissue of interest is isolated. The obtained mRNA is increased in copies by following methods:
Purification of mRNA by chromatography- in this method oligo-dT column is used which retains mRNA molecules, resulting in their enrichment.
Density gradient centrifugation method- this method is used to spin down mRNA.
A string of 50 – 250 adenylate residues (poly A Tail) is present in the 3′ ends of eukaryotic mRNA, which makes the separation easy from the much more prevalent rRNAs, and tRNAs using a column containing oligo-dTs tagged onto its matrix.
mRNAs bind to the column due to the complementary base-pairing between poly (A) tail and oligo-dT when an cell extract is passed through oligo-dT column. In an unbound fraction ribosomal RNAs and transfer, RNAs flow through. A low-salt buffer is used to elute the bound mRNAs.
First and second strand of cDNA synthesis:
As, mRNA is single-stranded it cannot be cloned as such and is not a substrate for DNA ligase. It is first converted into DNA before insertion into a suitable vector, which can be achieved using reverse transcriptase (RNA-dependent DNA polymerase or RTase), obtained from avian myeloblastosis virus (AMV).
Annealing of a short oligo (dT) primer to the Poly (A) tail on the mRNA takes place.
Enzyme, (Reverse transcriptase) extends the 3´-end of the primer using mRNA molecule as a template producing a cDNA: mRNA hybrid.
By RNase H or Alkaline hydrolysis mRNA from the cDNA: mRNA hybrid can be removed to give a ss-cDNA molecule.
Cloning of cDNAs:
Vectors generally used to clone cDNAs are phage insertion vectors. The advantages of using Bacteriophage vectors over plasmid vectors are as follows;
When a large number of recombinants are, required bacteriphage vectors are more suitable for cloning low-abundant mRNAs as recombinant phages are produced by in vitro packaging.
As compared to the bacterial colonies carrying plasmids, bacteriophage vectors can easily store and handle large numbers of phage clones. Particularly in the isolation of the desired cDNA sequence involving the screening of a relatively small number of clones, plasmid vectors are used extensively.
Applications of cDNA libraries/cloning:
To discover novel genes.
Gene functions in vitro studies by cloning full-length cDNA.
To determine alternate splicing in various cell types/tissues.
Various non-coding regions from the library can be removed with the help of cDNA libraries.
For detection of the clone or the polypeptide product, gene expression is required and is the primary objective of cloning.
To study the expression of mRNA.
Disadvantages of cDNA libraries:
Parts of genes found in mature mRNA are present in cDNA libraries.For example, (those involved in the regulation of gene expression), will not occur in a cDNA library which are sequences before and after the gene.
For isolating the genes expressed at low levels, cDNA library cannot be used, as there will be very little mRNA for it in any cell type.
Applications and uses of cDNA library:
Due to the removal of non-coding regions, storage of reduced amount of information can be possible.
In prokaryotic organisms, cDNA can be directly expressed
In reverse genetics, cDNA libraries are useful, where the additional genomic information is of less use.
Gene coding for particular mRNA can be isolated using cDNA library.
cDNA library and Genomic DNA library differences:
Non-coding and regulatory elements found in genomic DNA are absent in cDNA library.
Detailed information about the organism can be obtained using genomic libraries, but are more resource-intensive to generate and maintain.
Construction and applications of genomic library
Introduction:
An organism specific collection of DNA covering the entire genome of an organism is considered as a genomic library. DNA sequences such as expressed genes, non-expressed genes, exons and introns, promoter and terminator regions and intervening DNA sequences are present in a genomic library.
Some common steps in construction of a genomic DNA library includes; purification and fragmentation of genomic DNA, isolation of gDNA followed by cloning of the fragmented DNA using suitable vectors. Protease and organic (phenol-chloroform) extraction is used to digest eukaryotic cell nuclei. Thus, the genomic DNA obtained is too large to incorporate into a vector and needs to be broken into desirable size fragments. Physical methods and enzymatic methods are used for fragmenting DNA. Representative copies of all DNA fragments present within the genome can be collectively obtained in a genomic library.
Mechanisms for cleaving DNA
(a)Physical method
In this method, genomic DNA is sheared mechanically using a narrow-gauge syringe needle or sonication to break up the DNA into suitable size fragments that can be cloned. About 20 kb fragment is desirable for cloning into Lambda based vectors. Variable sized DNA fragments may result due to random DNA fragmentation. Large quantities of DNA are required in this method.
b) Enzymatic method
In this method, restriction enzymes are used for the fragmentation of purified DNA.
The action of restriction enzymes will generate shorter DNA fragments than the desired size and hence this method is limited by probability distribution.
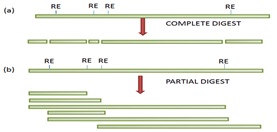
Multiple recognition sites for a particular restriction enzyme are present in a gene to be cloned; the complete digestion will generate fragments that are generally too small to clone and hence the gene may not be represented within a library.
Partial digestion of the DNA molecule is usually carried out using known quantity of restriction enzyme to obtain fragments of ideal size, to overcome the problem of multiple sites.
Type of ends (blunt or sticky) are the two factors that governs the selection of restriction enzymes to be used and are generated by the enzyme action and susceptibility of the enzyme to modification of bases like methylation (chemical modification) can inhibit the enzyme activity.
Agarose gel electrophoresis or sucrose gradient technique is used to generate fragments of desired size and then ligated using suitable vectors.
(Figure –The complete (a) and partial (b) digestion of a DNA fragment using restriction).
Cloning of genomic DNA:
There are different types of vectors available for cloning large DNA fragments. λ phage, yeast artificial chromosome, bacterial artificial chromosome etc, are suitable vectors for larger DNA and λreplacement vectors like λDASH and EMBL3 are preferred for construction of genomic DNA library. Selected DNA sequence into the vector is ligated by using T4 DNA ligase.
Advantages of genomic libraries:
It is used to identify a clone encoding a particular gene of interest.
Prokaryotic organisms having relatively small genomes can be mapped using genomic libraries.
Genome sequence of a particular gene, including its regulatory sequences and pattern of introns and exons can be studied from Genomic libraries from eukaryotic organisms.
Genetic load can be defined as the reduction in average or mean fitness of the population due to deleterious alleles (lethal alleles) which may cause death or sterility of the affected genotype.
The term genetic load was first used by American geneticist H.J Muller.
Genetic load is given as:
Equations used to establish genetic load consider single locus systems with alleles. (L= wmax – w / wmax))(1). Wmax is hypothetical fitness.
L= 1-w(2) where w, is the average or mean fitness of population.
{P2w1 + 2pqw2 + q2w3} (where w1, w2 and w3 are relative fitness of the genotype AA, Aa and aa.)
Factors affecting Genetic load: Selection, Mutation, Balance/segregation and heterozygote advantage.
Types of genetic load:
Mutational load:
Genetic load arising due to deleterious alleles caused by mutations is known as mutational load.
If a recessive gene is deleterious in homozygous condition, the loss in frequency of individuals incurred by genetic load= sq2
Suppose, if N individuals were in a population before selection than sq2 * N are eliminated because of genetic load (genetic deaths).
Therefore, recessive load caused by deleterious recessive allele is given as
L= sq2= u (mutation rate)
For a dominant deleterious allele, loss in frequency of individuals due to genetic load is:
Frequency of affected individuals * selection co-efficient, that is
=2p * s
=2 * u/s*s= 2u
Therefore, L= 2u.
Segregational/balanced load:
Due to heterozygote advantage, as the frequency of heterozygote’s increases, homozygotes are produced that die either before birth or are sterile.
Therefore there is wastage of alleles or genetic cost which we call as genetic load.
The balanced load gets created during selection, favouring allelic or genetic combinations which form inferior genotypes every generation by segregation.
In case of heterozygote advantage, mean fitness of population is, w= 1- sp2– tq2.
AA
Aa
aa
Initial zygotic frequency
P2
2pq
q2
Fitness
(1-s)
1
(1-t)
Each genotype contribution to next generation
P2(1-s)
2pq
q2(1-t), q2– sp2– tq2
Allele frequencies are as follows:
P’= t/s+t and q’= s/s+t, (equation m), where ‘s’ and ‘t’ are selection co-efficient against AA and aa genotype respectively. Substituting the (equation m) allele frequencies in equation frequency of w, we get: w= 1-sp2-tq2
= 1-s(t/s+t)2 – t(s/s+t)2
= 1-st2-ts2/(s+t)2 = 1-{st(t+s}/(s+t)2(so, here t+s and the square gets cancelled)
Therefore, w= 1-st/s+t and segregational load is L= st/s+t.
The above equation is applicable to one locus. For several loci the segregational load will increase dramatically.
Since population fitness will decline rapidly with increase in number of loci, thus:
W= (1-L)ihere i, is no of loci showing over-dominance.
The required increase in the reproduction rate of surviving individuals will be quite large.
In most species, however the possible increase in the reproduction rate per surviving individual will be usually much smaller than required to compensate for this genetic load.
As a result, the population would most likely become extinct over time.
Application and Significance:
Theory known as Fitness indication theory was build on the assumption that in humans, deleterious germ line mutations can influence fitness outcomes related to their pleiotropic effects on traits.(can influence different or multiple traits at once). There are assumptions and predictions that there exists a fitness factor(F) among traits that signals sensitivity of development to perturbation stemming from deleterious mutations that are present. Therefore, among sexually reproducing taxa, there should exist genetic correlation between distinct traits. The theory was proposed in the year 2000 by Houle and Miller.
In future micro-evolutionary trends, in particular for fitness stemming from purifying selection and from mutation accumulation in industrialised population is significant in terms of concept of genetic load.
Genes may be present on the same chromosome or different chromosome. Many genes are present one chromosome.
Characters controlled by genes express in next generation, when the genes are present on different chromosome. Here, we can say they assort independently as per the law of independent assortment given by Mendel.
Genes tend to inherit, if they are present on the same chromosome and are close to each other.
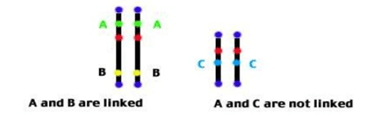
Thus, linkage can be defined as coexistence of 2 or more genes on the same chromosome.
Extensive work carried out in drosophila led to the discovery of term Linkage, which was given by Thomas Hunt Morgan.
On a given chromosome, if all the genes are located Physically, than it is called as Linkage group.
Characteristic of Linked Genes:
1:1:1:1 test cross ratio is observed when genes assort independently on different chromosomes.
Linked genes remain in the same combination as they are in parents, and do not assort independently.
Chromosome Theory of Linkage:
It was given by Castle and Morgan.
During the inheritance process, linked genes remain attached to chromosomal material and are present on the same chromosome.
Strength of linkage is determined by the distance between linked genes. More stronger linkage is exhibited by closely related genes when compared to widely located genes, which has weaker Linkage.
In a chromosome, genes are arranged in linear fashion.
Types of linkage:
Basically two types of linkages are found based on works in drosophila by Morgan and co-workers.
Complete Linkage:
In this linkage, parental characters appears together for more than two generations in a continuous fashion.
Genes transmit together and remain close to each other in this type of linkage.
Example:
Fourth chromosome mutant of Drosophila Melanogaster, exhibits complete linkage carrying genes for bent wings (bt) and shaven bristles (svn).
Between 2gene pair, there is absence of independent assortment indicating very strong complete linkage.
Incomplete Linkage:
Widely located linked genes on chromosomes which are capable of crossing over are called as incomplete genes and the pattern of their inheritance is termed as incomplete linkage.
As homologous non-sister chromatids(one part of chromosome) exchange varied length fragments during meiotic phase, the linked genes do not always stay together.
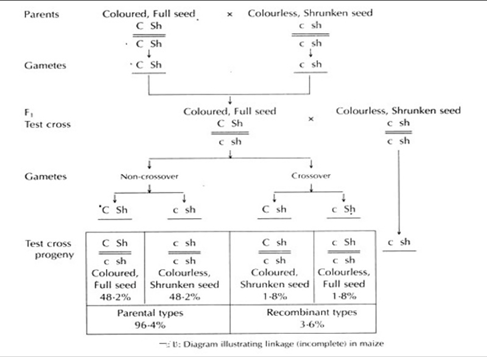
+Example– The phenomenon is observed in maize, female drosophila, tomatoes, pea, mice, man and poultry. Incomplete linkage in Maize, was studied by Hutchinson, where he observed alleles for incomplete linkage between colour and shape of the seeds.
Chromosomal Mapping:
Genetic maps: It refers to representing the relative distance between linked genes in a diagrammatic manner.
It is also called crossing-over map, as it is the outcome of crossover.
Chromosome mapping is construction of genetic maps for different chromosomes.
Steps involved in construction of chromosome maps:
Crossing over frequency between 2 genes is directly proportional to the distance between them on the chromosome.
One unit map distance between genes(1%frequency of Crossing over between 2genes) is called as Centimorgan.
Step 1: Conduct hybridization experiments(among wild and mutants) to know number of genes and chromosomes of a particular species.
Step2: Determine the relative distance between linked genes after knowing the linkage groups.
Step3: calculate 2gene distance based on the percentage of crossing over.(crossing over directly proportional to gene distance).
Example: Map distance between 2 linked genes is one centimorgan, if percentage of cross over between 2 linked gene is 1 percent.
Hypothetical Construction of chromosomal map:
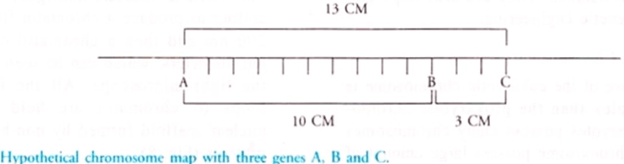
Assume that there are 3types of genes: A, B and gene C.
Say 10% is the cross over percentage between A and B, they can be plotted on a linear scale.
3% say, is the cross over percentage between B and C, than cross percentage between A and C will determine position of C. It can be away from A or 3units from B.(It can be interpreted by crossing A and C).
Now suppose say, 13% is the cross over percentage between A and C, than C will be plotted 3centimorgans right side to that of gene B.
Formula to determine Recombination frequency:
It can be given as: (Recombination frequency= Total no of recombinants/ Total no Progeny)
Significance of mapping Chromosome:
Exact location of genes in a Chromosome can be determined.
Approximate distance and correct order between the genes can be obtained and is useful for Genetic studies.
Chances of crossing over between genes and their linkage can be studied and obtained using mapping technique.
To identify in which loci of chromosomes exactly genes are present.
Used in genetic manipulation studies.
This chromosomal mapping proves beneficial in Autosomal Dominant testing, which helps to understand the ancestor history by knowing which DNA segments came from which ancestor.
The sudden inheritable changes in the genetic materials are called mutations.
Mutations may be harmful, beneficial or neutral in their effect. Majority of mutations are harmful, because most of the organisms are already adapted and any changes would be disadvantageous. But some mutations are beneficial.
CLASSIFICATION OF MUTATION
1. GENE MUTATION OR POINT MUTATION
These are changes that occur in the fine structure of the genetic material. These changes are also heritable. Usually it involves a single nucleotide or nucleotide pair.
2. CHROMOSOMAL MUTATIONS
These are changes that affect larger regions of a chromosome and the number of chromosomes. Such changes in the structure and number of chromosomes can be observed under microscope. It is associated with the appearance of new traits in organisms. Five types of chromosomal mutation:
Deletion-due to breakage apiece of chromosome is lost
Insertion-chromosomes segment breaks off, flips and reattaches.
Duplication-Gene sequence is repeated.
Translocation-Part of one chromosome is relocated with another chromosome.
Non Disjunction-Gametes will contain too many chromosomes due to the failure of separation during meiosis.
TYPES OF MUTATIONS
Somatic and Germinal Mutations: If mutation occur in the somatic cells (non-reproductive cells) it is called somatic mutation. If a mutation occurs in the reproductive cells it is called germinal or genetic mutation.
Dominant and recessive mutation: when a mutation produces a dominant phenotypic expression it is called dominant mutation. If a mutation produces a recessive expression, it is called recessive mutation.
Micro and macro mutation: When mutation produces small visible changes, e.g., white ye in Drosophila. Such mutations are called micro mutation. Mutations which produce prominent visible changes, e.g., albino maize, Ancon Sheep etc. are called macro mutations.
Spontaneous and Induced Mutations: Naturally occurring mutations are called spontaneous mutations. Mutations produced artificially by action of certain agents are called mutagens. Such mutations are called induced mutations.
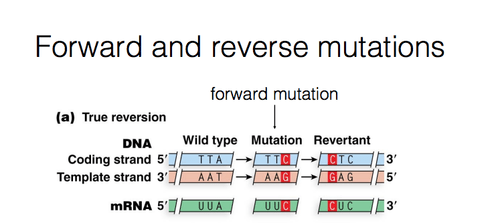
Forward and reverse mutations: During mutation, a change take place from the normal type (wild type) to a new form (mutant form).This is called Forward mutation. The mutants may revert to the wild forms. Such changes are called reverse or back mutations.
Lethal mutations: Some mutations affect the vital functions of an organisms. This leads to the death of an organism .Such mutations are called lethal mutations.
MUTAGENS
Mutagens are substances which induce mutation. Such mutations are called Induced mutation. The frequency of such mutations is higher than spontaneous mutations. Mutagens are classified into two major groups:
1. High Energy Radiations
2. Chemicals
Applications of Gene Mutation
G.J .Mendel could not have put froth the basic ideas on inheritance, had all the pea plants were only tall etc.
T.H Morgan was able to evolve his ideas about sex-linked inheritance only when he could find out the mutant white-eyed Drosophila flies.
T.Benzer could analyses the fine structure of genes with the help of mutant forms of T4 phages.
Plants with new and beneficial traits have been evolved through induced mutations. Plant breeders have produced mutants in barley, wheat, oats, soybean, tomatoes etc., which have desired characters like increased yield and resistance to disease.
The yield of Penicillium has been increased through mutation.
Genes of an organism is encompassed by Genome. Gene is a hereditary unit, which consists of all the nucleotides and forms a part of chromosome. Spatial distribution of chromatin(DNA and protein) within a cell nucleus is called as Nuclear Organisation. There are different levels in nuclear Organisation.
History:
Chromosome organisation into distinct regions within a cell nucleus was first observed and discovered in the year 1885 by Carl Rabl. With advancement in microscopy technologies, in the year 1909, chromosome territories were discovered by Theodor Boveri when he observed that chromosomes take up separate nuclear regions.
Prokaryotic Organisation:
Not well defined nucleus is not present in Prokaryotes. Prokaryotes means, ‘before nucleus’. Chromosomal DNAin prokaryotes is present in structures called Nucleoid.
Circular DNA is present in loops or domains in the nucleoid binded to scaffold proteins which are attached to cell membrane.
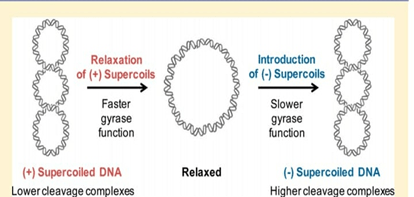
Bacterial chromosomes are huge, but still it gets fitted inside the cell. This occurs because of Supercoiling of DNA.
DNA Supercoiling:
Prokaryotes, compress their DNA into small pieces by supercoiling.
In bacterial genome the circular DNA is supercoiled because of addition of turns in the double helix structure of DNA.
Two enzymes are necessary for supercoiling in prokaryotes, Topoisomerases(type 1) and DNA gyrase.
Chromosomal DNA is compacted to 1000 folds in order to fit DNA within bacterial cells.
DNA is organised into loops by proteins and further this loops compact the DNA by 10 folds.
Supercoiling folds DNA by additional 100 folds.
Supercoiling of DNA can be negative or it can be positive. Negative supercoiling takes place in the opposite direction to the double helix.
Positive supercoiling is when twisting occurs in the same direction as that of double helix DNA.
During normal growth, most of the bacterial genomes are negatively supercoiled.
Proteins involved in Supercoiling:
Multiple proteins are involved in DNA folding and condensation, which was reported in the year 1980s and 1990s.
One of the most abundant protein in nucleoid, HU, binds DNA with the help of enzyme Topoisomerase I, introducing sharp bends in the chromosome, generating tension which is necessary for negative supercoiling.
One more protein called Integration Host factor(IHF) binds to specific genome sequences and produces additional bends. This was showed by Rice et.al in the year 1996.
Topoisomerases and DNA gyrase maintains the supercoiling, once the genome gets condensed.
Genes involved for modulating response to environmental stimuli can be altered in terms of their expression patterns, and this is done by H-NS which is a maintenance protein during transcription.
How these tightly packed genes gets accessed by proteins?
Nucleoid in Prokaryotes appears as irregular mass, but when cell is treated with chemicals for inhibiting transcription or translation, Nucleoid becomes Spherical.
Small regions of chromosomes project out during transcription process from nucleus to cytoplasm. In this region they unwind and gets associated with Ribosomes and thus allows easy access for various different types of transcriptional proteins.
Eukaryotic Organisation:
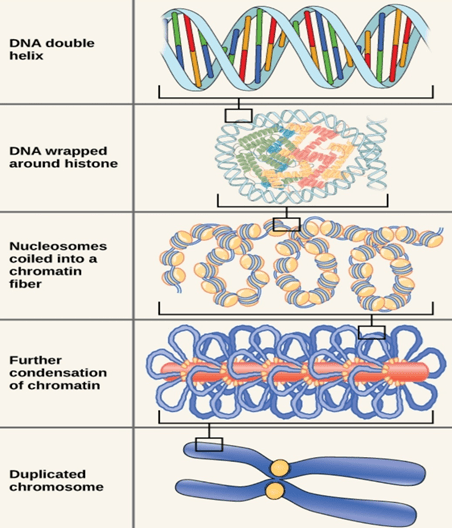
Large amount of eukaryotic DNA is packed in chromosomes present within nucleus. DNA and proteins together constitutes a chromosome.
Mostly histone proteins are present in abundance, but certain other proteins are also present in less amount termed as Non- histone proteins.
DNA-protein nuclear complex is called as chromatin.
Length of packaging varies, for example in humans 1.6cm shortest DNA molecule can be wrapped and as large as 8.6cm can also be wrapped.
Packaging of DNA into chromosomes involves folding and has different stages involved in it.
Stage 1(Nucleosome):
Chromosomal DNA binds to histone proteins in first stage of organisation.
DNA wraps around histone octamer(2copies of dimers and tetramers each) at regular intervals. DNA-histone complex is called the chromatin.
DNA wrapping around histone proteins forms bead like structure which is called Nucleosome.
Nucleosome core particle wrap DNA for about 1.67 left handed turns and contains 146base pairs of wrapped DNA.
DNA that connects to Nucleosome is called as Linker DNA.
DNA in this form is seven times shorter when compared to double helix structure without histone proteins. The size of beads is 10nm in diameter compared to double helix which is 2nm.
Histones are: H1, H2a H2b H3 and H4. H1 is not included in core histone and it connects to linker DNA.
Histone proteins are basic proteins mostly made up of lysine and arginine amino acids which are positively charged.
Stage 2(30nm fibre):
In this level of compaction, linker DNA and Nucleosome are coiled into a 30nm fibre.
Because of this coiling, the chromosome further gets shortened and becomes 50 times more shorter than the extended form.
Fibre is formed when H1 histone binds to linker DNA at each Nucleosome. When H1 histone interacts with each other, nucleosomes gets pulled together.
Stage 3 (Radial Loops):
When chromosomes are deprived of histones, they possess a central fibrous protein scaffold, to which DNA gets attached in loops.
The main role of these fibrous proteins is to make sure that each chromosome in non-dividing cell occupies a particular area of nucleus that doesn’t overlap with other chromosomes.
GM Plants: To prepare a GM plant, new DNA is transferred into plant cells. Genetic engineering techniques are used to modify DNA. Genetic composition of the plant is altered by adding specific useful genes. Once the new DNA is inserted, than the cells are taken and grown in tissue culture using appropriate medium where they develop into new plants and will inherit the new DNA which was inserted.
History:
Tobacco plant, first genetically modified plant was produced in the year 1982. These plants were modified for antibiotic resistance. The first country to commercialise use of transgenic plants was China. The tobacco plants were made antibiotic resistant by creating a chimeric gene which joined the antibiotic resistant gene present on the T1 plasmid of Agrobacterium. Bt cotton was the first commercialised genetically modified crop used in India which was made by Maharashtra hybrid seeds company along with Monsanto company from USA.
Process of developing GM Plants/Crops:(6 steps)
Isolating Gene of interest- Gene of interest is isolated from other plants or organism. Information like structure, function and location of chromosomes is useful in identifying gene of interest in an organism. Information about gene function and it’s regulation in donor organism(organism from which gene is taken) and in recipient organism(organism in which the gene is inserted) must be known fully before starting the experiment in order to minimize the adverse effects.
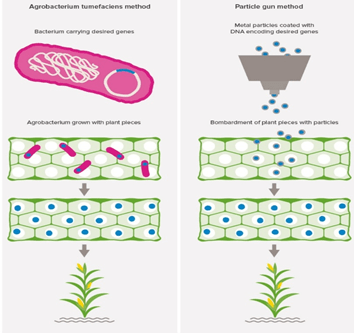
Gene insertion by using Transfer vector and plant transformation- Plasmid from Agrobacterium Tumifaecins, are used as common transfer tools. Using rDNA technology, the gene of interest is inserted into the plasmid. Plant cells or explants are mixed with Agrobacterium cells containing the plasmid with new genes. T-DNA, piece of plasmid is taken up by the cells. Desired genes are inserted into one of the plant chromosome by Tumifaecins, and now the plant is called Genetically modified. Other method used for transfer is Gene gun method or particle bombardment method. In this method, the relevant metal surfaces are coated with small DNA fragments and these particles are bombarded into plant cells. Mostly Tungsten or gold particles are used for coating DNA (called microprojectiles). This method is a bit costly but can be effective.
Selection and Regeneration of modified plant cells to form a whole plant: Generally, a small fraction of plant cells take up the gene of interest after transformation. Hence, selectable marker genes, that favours antibiotic and herbicide resistance are used to favour growth of transformed cells. After the process, the transformed cells are regenerated into a whole plant using plant tissue culture method.
Plant transformation verification: The gene inserted has to inherit normally, so this needs to be verified. For this purposes, tests are performed to determine number of copies inserted, intactness of the copies inserted and effects of inserted gene with the other genes. In this, gene expression (mRNA-proteins) is also checked to confirm whether gene is functional or not.
Testing of plant performance: checking plant performance is vital. After transformation, only fraction of plant cells have the copies of inserted gene. So selective markers are used to favour growth of transformed cells. The resistance genes along with genes for desired traits are transferred using a suitable vector. So when cells are exposed to antibiotics or herbicides, only transformed cells with this selective markers will grow. This way performance is checked and only transformed cells are taken to regenerate and create a whole plant by using tissue culture.
Safety assessments: safety assessments are necessary in terms of food and environment. There are different tests to determine whether the released modified plant is safe for consumption or for cultivation to produce higher yields without damaging the environment.
GM Crops in India:
Bt cotton:
Bt cotton was developed to tackle the boll worm infection in cotton plants. The Bt cotton variety was developed by Maharashtra hybrid seeds company along with Monsanto, USA.
GEAC(genetic engineering approval Committee), in 2002 approved Bt cotton making it the first genetically modified plant in India to receive the approval.
Bt cotton is an insect resistant genetically modified crop.(made resistant to cotton boll worms, that destroys cotton plants).
Bt is a protein from bacillus thureingenesis, bacteria which has 200 different types of Bt toxins. Each toxin affects and works on different types of insects.
Cry group of endotoxins in Bt cotton are modified by inserting a gene with toxin crystals, when organisms(insects) ingests this genes with toxins, toxins dissolves the gut lining of the insects leading to its death. Thus, the crop plant is protected from boll worms.
Bt Brinjal:
It was developed to give resistannce against lepidopteran insects, specifically to leucinodes orbonalis, which is a fruit and shoot borer in Brinjal plants.
Bt Brinjal was developed by Maharashtra hybrid seeds company in collaboration with Tamilnadu agriculture University and Dharwad institute of agricultural sciences.
GEAC approved commercialisation of Bt Brinjal in the year 2007, however , due to lack of proper safety and efficacy and lack of scientific consensus, it was banned in the year 2010.
HT Mustard:
DMH(Dhara Mustard) was created to reduce the demands of edible oil imports of India.
It was created by Delhi University professor, Deepak Pental.
DHM-11 was created using transgenic technologies in particularly involving Barstar/Barnase gene technologies.
Male fertility is conferred by Barnase gene, while Barstar, restores fertile seeds producing abilities of DHM-11.
GEAC approved it in the year 2017.
Advantages of GM crops:
Crop Protection: Resistance to diseases, pests, insects and herbicides. Resistance is achieved by using Genetic engineering methods and by using toxins in case of Bt cotton.
Economic Benefits: GM plants increases the yield double times compared to the normal plants.
With increasing demands of quality food, GM crops can be beneficiary in providing and supplying food at much faster rates.
Concerns with GM crops:
Health concerns: Transfer of antibiotic resistance markers or allergens are some of the potential risks. Example- In HT Soya areas, in Argentina, birth defects and childhood Cancers were increased by threefold. This was a report based study.
Environmental concerns: can reduce diversity of species. For example, if insects that are not be killed gets killed by developing modified crops can reduce species diversity. Super weeds(transfer of genes from one crop to other creates super weeds) which are resistant to most of common control methods.
Economic concerns: Launching of GM crop to market is costly and time consuming process. Also violation of ethical issues have been raised as a concern, for example- organisms intrinsic natural values have been violated by mixing it with other species.
GEAC(Genetic engineering approval Committee)– It was approved under ministry of environment, forest and climate change for manufacturing, use, import, export of GMOs (genetically modified organisms). This committee is also responsible for giving technical approvals for proposed GMO products including field trials.
Safety of GM crops and it’s related products is monitored by Institutional Biosafety Committee (IBSCs).
GM Animals:
GM animals can be created by inserting a foreign gene of interest into their genomes. rDNA technology is used for construction of foreign gene. Along with gene, DNA is also modified and contain different sequences in order to incorporate and express into the host cells.
History:
Mice embryos in-vitro manipulation was first reported in the year 1940 using a culture system. In Angora Rabbits, first successful transfer of embryos was achieved in the year 1891 by Walter Heape. Modern genetic modifications began in the year 1973, when Herbert Boyer and Stanley Cohen first discovered and demonstrated that gene from one organism can be cut, and pasted to other organism. Mouse is the first genetically modified animal which was developed by Rudolph Jaenisch, in the year 1974.
Examples of GM Animals:
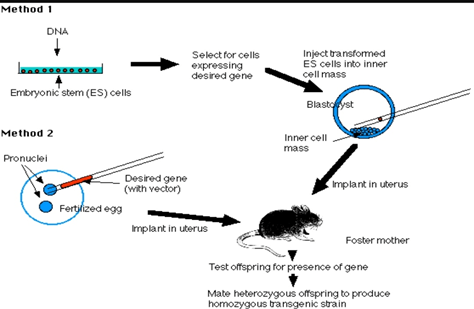
Mice: GM mouse models are used extensively as models for studying and understanding different diseases. Two methods are well known for developing GM mice, Embryonic stem cell method and Pronucleus method.
Embryonic stem cell Method: (ES)(method 1)
Mouse blastocyst has inner cell mass from which ES are harvested.
rDNA technology is used to make the DNA containing the desired gene of interest, vector and promoter and enhancer sequences.
ES cells are transferred in culture. When ES cells are exposed to DNA, it gets incorporated into it.Transformed cells are selected and injected into inner cell mass of mouse blastocyst.
Transfer of embryo. Pseudo pregnant mouse is created and the embryo is transferred into its uterus.
Offspring produced by it is tested. For testing, remove small piece of tissue from tail and examine it’s DNA. It should be present in 10-20% and it should be heterozygous for the gene.
Transgenic strain is established by mating two heterozygous mice and screening their offspring’s.
Pronucleus method:(method 2)
rDNA technology is used for preparing DNA with desired gene, vector and promoter and enhancer sequences.
Freshly fertilized eggs are harvested before sperm head becoming Pronucleus.
The male Pronucleus is injected with DNA that is prepared.
Zygote formed by pronuclei fusion is allowed to divide by mitosis to form 2 cell embryo.
The embryo is than implanted into pseudo pregnant foster mother.
Than the rest of the steps are common with respect to ES method. Offspring test is performed followed by establishing Transgenic strain.
GM chicken:
Embryos are infected with viral vectors carrying human gene with a therapeutic protein and promoter sequences.
Human gene is transformed with the rooster sperm or appropriate promoter.
Check for transgenic offspring’s..
The method is cost effective.
C. GM Sheep’s:
Connective tissue cells of sheep are treatedwith a vector, which has 2 homologous regions to that of COL1a1 gene of sheep, alpha 1 Anti-trypsin coded by human gene, antibiotic neomycin resistant gene, beta lacto globulin gene promoter site and ribosome binding sites for beta lacto globulin to be translated.
Transform the cells and fuse with enucleated(without nucleus) sheep cells.
Next step is implantation into uterus of female sheep(called ewe).
Lambs produces large amounts of milk when treated with hormones.
However, this method implemented by one of the company in 2000, abandoned it in 2003 because for purification of protein from sheep’s milk, the cost of expenses were almost doubled.
Applications of GM Animals:
GM animals are used as models to understand the disease process and it’s progression.
They can be used as models to test new therapeutics that are being developed for treatment of diseases.
GM animal models are also used to study gene function. For example- animals with certain genes being turned off or non functional genes can be studied to understand how turning off of this genes can lead to diseases and the mechanisms behind it.
Can be used in agriculture to confer resistance to diseases against pathogens.
Knockout mice- used extensively in research to understand genes for which mutant strains are not available.
Knock-in mice: It removes certain DNA sequences that otherwise blocks transcription. So the target gene can be turned on as per wish. Also new gene can be introduced by replacing one of the mouse gene.
Molecular marker is a sequence of DNA in the genome that can be traced and identified. RAPD is a molecular marker that helps to identify genetic variations. Single arbitrary primer is used in RAPD.
History:
It is used to identify nucleotide polymorphism. This method came into existence during the year 1990 when William et.al first used this approach.
Principle:
Shorter oligonucleotide primers binding to different loci is used for amplification of random sequences from complex DNA template.
The amplified PCR product depends on length and size of primer and the target genome.
RAPD is a PCR based method. All the components of PCR are required to perform RAPD like DNA template, dNTPs, reaction buffer, Mgcl2(cofactor), arbitrary primer, Taq polymerase.
First step basically involves denaturation, where the PCR products are kept at higher temperature of 94 degree Celsius.
Temperature is lowered to 40-65 degree Celsius in annealing process where the primer binds to the target DNA sequence.
Further Taq polymerase adds DNA-hybrid primers to 3’ end and extends to the other end of target sequence.
The process is repeated until complete replication of amplified product is achieved.
Outline of RAPD Analysis:
Genomic DNA is isolated.
The isolated DNA is denatured.
DNA template is annealed with primers- Annealing.
Complementary strands of DNA are synthesized after the extension phase.
Gel electrophoresis is used to identify amplified products.
Procedure of RAPD:
Materials Required:PCR components, RAPD arbitrary primers, gel electrophoresis reagents and equipments, Eppendorf’s, PCR tubes, micro centrifuge, micropipettes, deep-freezer, UV transilluminator connected with a system.
Master mix is prepared(control and sample) with all the PCR components.(thaw(keep it in ice) and vortex).
The master mix is aliquoted in PCR tubes.
DNA templates is added in the PCR tubes.
The tubes are placed in allotted blocks in the PCR thermocycler machine.
The reaction setting is adjusted and different temperatures are set for different cycles.
Ideal reaction is: step 1 for 94 degree Celsius (1min), step 2 for 35 degree Celsius (1min) and step 3 at 72degree Celsius for 5min. For final step, temperature is again 72 degree Celsius for 5min.
DNA loading dye is added to the amplified product and Agarose gel electrophoresis is performed. (% of agarose gel is determined based on sample size).
The gel is visualised under UV trans illuminator connected to a system.
Applications of RAPD:
As molecular markers in plants: Using RAPD, genetic maps are constructed in plants. For example- In coffee 15 linkage groups are constructed using RAPD marker.
Desirable trait can be selected directly using RAPD. This marker can be screened with the desired trait anytime during breeding programs. This gives breeders advantage, as they can trace the desired trait.
RAPD is widely used to identify certain genes which are resistant to diseases. For example- In barely crop, the gene rp94 is resistant to stem rust.
Polymorphism studies can be done using RAPD.
RAPD markers are used in evolutionary and population genetics for identifying genetic variations among different species.
Used in germplasm(genetic material of Germ cells) characterization.
RAPD can be employed to detect somaclonal variations, variations observed in somatic cells of regenerated plants.
RAPD PCR is used for detection of genetic polymorphism in leishmania strains(parasites causing leishmaniasis). Example- This type of test was successfully performed in Tunisian patients.
Merits of RAPD:
For designing specific primers, no DNA probes and specific sequences is required.
It is a quick, simple and efficient method as it does not involve blotting or hybridization steps.
Amount of DNA required in the process is small compared to other methods.
Number of fragments are higher.
Compared to other assays, market cost for RAPD is relatively low.
Demerits of RAPD:
Since RAPD markers are Dominant, it will be difficult to distinguish whether a particular DNA sequence is amplified from heterozygous locus or homozygous locus.
RAPD is exclusively laboratory based technique and hence requires all the necessary components and suitable PCR conditions to obtain desired result.
This method is sensitive to changes in DNA, PCR components and PCR conditions and hence has problems with reproducibility.
Null alleles ( it is a non-functional allele caused due to mutations) cannot be detected directly by this method.