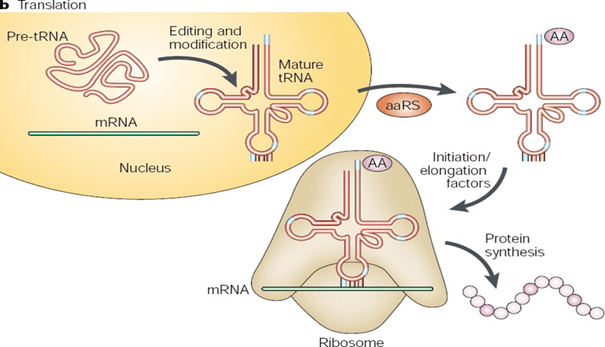
BY: RAHUL ANDHARIA (MSIWM001)
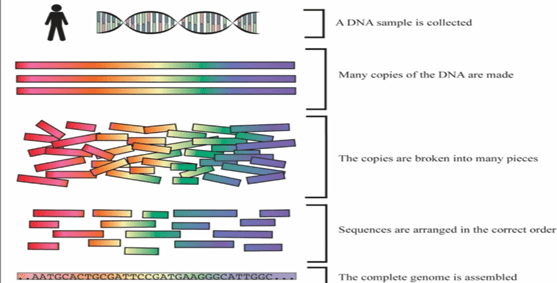
Technique to determine the nucleotide sequence at certain portions of DNA, which is unique to all is known as DNA fingerprinting. Human DNA is only 0.1% unique and 99%similar. This 0.1% makes every individual unique. DNA remains stable even after 1000 years. DNA Fingerprinting can also be called as DNA profiling. Sample of DNA can be collected and series of procedures are performed to determine the amount of polymorphism(difference in phenotypic expression)in noncoding repetitive sequence.
History:
By using Restriction digestion length polymorphism, in the year 1985, the concept of DNA fingerprinting was given by Alec Jeffery. He used Autoradiography as well as RFLP( Restriction fragment length polymorphism) methods for his work. DNA Fingerprinting work in India was revolutionized by Dr Lalji Singh, and hence he is known as father of DNA fingerprinting in India.
Principle of DNA fingerprinting:
- 95% of the human genome is made up of noncoding, repetitive DNA, about only 5% is regulated by formation of proteins, we know it as genes.
- By using Density Gradient centrifugation, this region’s can be separated as satellites, and can be called as satellite DNA.
- Base repetition , is in tandem in the satellite DNA. Satellite DNA are of two types namely, microsatellites and minisatellites depending upon number of repetitive tandem units, base composition and their length.
- Polymorphism is shown by satellite DNA. Generally the term polymorphism is used when a variant at locus present with 0.01 frequency in a population.
- Mutations gives rise to variations. Non coding region has mutations and these mutations gets build up with time, and forms basis of DNA polymorphism.
- Length polymorphism, is the basis of the junk DNA region, showing changes in the Physical length of DNA molecule.
- Number of tandem repeated varies with specific loci. This repeats are of two types based on their size, STR(short tandem repeats) and VNTRs(variable number of tandem repeats).
- STRs have base repeats of 2-5bp while VNTRs has 9-80bp repeat.
- Number of VNTRs will differ at particular region of DNA primarily due to deletions, insertions or mutations in base pairs because child receives 50% of DNA from mother and the other 50% from father.
- Because of this reason, the VNTR component in each individual is different and this is the main principle in DNA fingerprinting concept.
- Thus, the basics of DNA fingerprinting is that, DNA of individuals upon restriction digestion, results in fragment sites which will differ in their cleavage sites positions.

General Steps involved in DNA fingerprinting:
- First step is isolation of DNA. This isolated DNA is than treated with chemicals to break open the cell membrane to remove other materials, and extract pure form of DNA. Sample is generally taken from blood, cheek swabs.
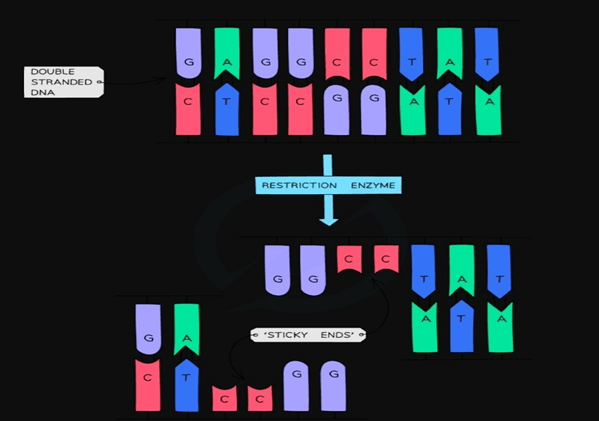
- The next step, is to digest the isolated DNA with restriction enzymes. Generally, repeated sequences(DNA portion with exact same sequence) are used so that they can be identified by same restriction enzymes. Common examples of DNA repeats used are VNTRs and STRs.
- As per the fragment size, digested fragments are than separated by using electrophoresis. (Separation based on size and charge by passing electric current).
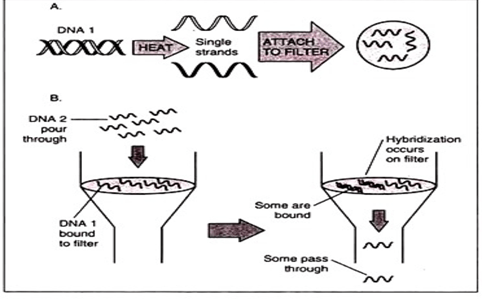
- Transfer (blotting) of separated fragment onto nitrocellulose of pvdf(polyvinyl difluoride) membranes. Gel is treated with weak acids, which break opens the DNA into individual nucleic acids which will easily get rub off onto paper.
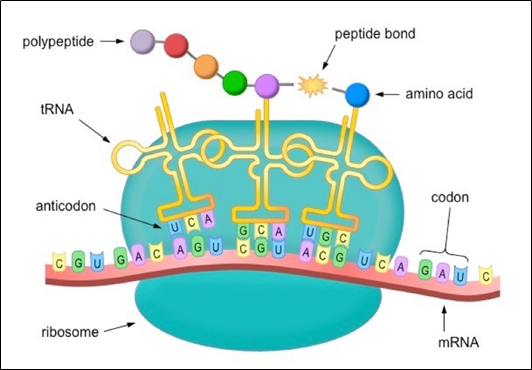
- Labelled VNTR probes are used to hybridize the fragments.
- Autoradiography, is used to analyse the hybrid fragments.
Methods of DNA fingerprinting:
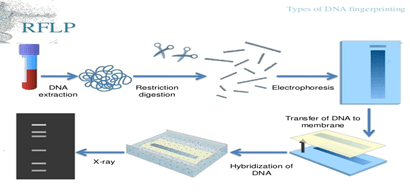
- RFLP (Restriction fragment length polymorphism):
- DNA isolation from sample material. Use large sample size for RFLP to obtain better results.
- After sample isolation, subject the DNA for restriction digestion.
- Separation of digested sample by Agarose gel electrophoresis.
- Transfer of separated DNA to nitrocellulose membrane. The sample is than hybridised by labelled probe which is specific for VNTR region.(southern blotting)
- After this, x-ray film is developed from the southern blot. On the x-ray film only hybridised regions(where the radioactive probe will bind) will be shown.
- Analysis with other samples and studying band pattern and comparing different samples will give the result of DNA fingerprinting.
Advantages: Gives more accurate results compared to the PCR method. Generally because, the sample size used is large, sample DNA is fresh and also there is no chance of amplification contamination, which tends to happen in PCR
Limitations: Takes longer time to complete and proves to be a costly method for more frequent uses.
- PCR Amplification of Short Tandem Repeats( STRs):
- Particular variable region is amplified in thousands of copies.
- Known repeat sequence of STR is amplified and separated with the help of electrophoresis.
- Distance travelled by STR is noted.
- Primers are specially designed for PCR amplification for the purpose of attaching highly conserved common non variable region of DNA which flanks the variable region of DNA.
- STR sequence sizes are compared with various samples to give the result of DNA fingerprinting.
Advantages: Requires small amount of sample, shorter duration, faster results and less costly method.
Limitations: chancesofamplification contamination, not very accurate method.

Applications of DNA fingerprinting:
Generally, this technique is employed to identify individuals of same species by comparing their DNA.
- Forensic science:
- DNA is isolated from the crime scene and is generally useful in solving crimes. The isolated DNA is compared with the VNTR prototype.
- Common materials used for profiling of DNA are: blood, hair, semen, body tissue cells, etc.
B. Paternity and Maternity determination:
- Dispute cases, inheritance cases and immigration cases are generally solved by parent-child VNTR analysis.
- VNTR are generally acceded from his or her parents to the individual.
C. Personal identification:
- DNA fingerprint pattern is unique in each individual.
- Hence, this is used as a personal identification bench mark and as a genetic barcode of identification.
D. In diagnosis of inherited disorders:
- Used in diagnosis of inherited disorders in new-born and prenatal.Some of this disorders include Huntington’s disease, haemophilia, cystic fibrosis, sickle cell anaemia.
E. Used to detect maternal cell contamination:
- Maternal cell contamination is the common problem with prenatal diagnosis. Sometimes the amniotic fluid contains maternal tissues.
- This type of contamination can increase the chance of false positive results and can hinder in carrier identifications.
- So by using STR or VNTR markers and by performing gel electrophoresis, maternal cell contamination is identified during genetic pregnancy testing.
F. Breeding program:
- Generally, breeders use phenotype to determine the genotype of plants or animals. It is difficult to determine whether plants are homozygous or heterozygous on the basis of appearance.
- Thus, DNA fingerprinting method can be used for precise determination of the genotype.
- This approach is used primarily in breeding hunting dogs and race horses.
DNA fingerprinting has many applications and is a useful technique in development of cures for inherited disorders, as the DNA prototype associated with disorder can be studied from the relatives. The accuracy for DNA fingerprinting relies more on RFLP method than in PCR methods.