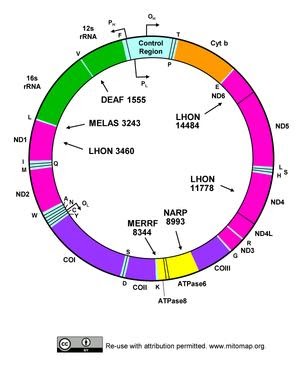
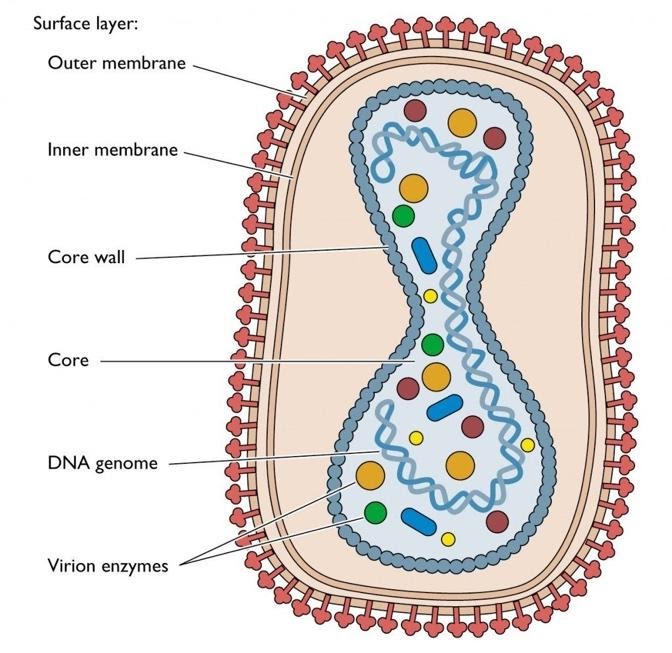
DNA within mitochondria was first detected in 1963. Human mitochondria represent the mammalian mtDNA. Human mtDNA is 16,569 bp, double stranded and circular. It codes for 13 polypeptides, belonging to OXPHOS family. mtDNA also codes for 22 tRNA,2 rRNA. It also has control noncoding regions. Various nuclear coded factors also known as precursor polypeptides are essential for the expression and maintenance of mtDNA. It was Sanger who found that circular mtDNA in vertebrates, has both Light Strand and Heavy strand. There are many differences considering nuclear DNA and mitochondrial DNA, which are as follows
MITOCHONDRIAL DNA
NUCLEAR DNA
Present in mitochondria
Present in nucleous
One cell contains 0.25% mtDNA
One cell has 99.75% n DNA
Mutation rate of mtDNA is 20 times faster than nDNA
Slow mutation rate
circular
Linear
1000’s of mtDNA copies/ cell
2 Copies / cell
Haploid
Diploid
Maternally inherited
Biparental inheritance
Replication repair mechanism is absent
Replication repair mechanism is present
Reference sequence by Anderson and Colleagues in 1981
Reference sequence in Human Genome Project in 2001
Size of genome is 16,569 bp
Size of genome is 3.2 billion base pair
The genetic code of nuclear DNA differ from mtDNA as such ‘TGA’ codes for tryptophan in vertebrate mitochondria, while it is a stop codon for nuclear DNA. ‘ATA’ codes for Isoleucine in cytosol, while it codes for methionine in mitochondria.
Mitochondrial Inheritance:
With most of the evidence provided, mostly there is maternal inheritance of mitochondria. Due to nucleotide imbalance and reduction in fidelity of polymerase γ, it causes higher mutation rate. This can be used as approach for human identity test, studying evolutionary and migration pattern.
mtDNA replication:
Factors for mtDNA replication:
DNA Polymerase γ is the polymerase enzyme, it is a heterotrimer with one catalytic subunit ( POLγA). POLγA has 3’-5’ exonuclease activity for proofreading. TWINKLE is DNA Helicase which unwinds double stranded DNA. mtSSB is binds with single stranded DNA to protect from nucleases. Vinograd and coworkers proposed the strand displacement theory, which emphasize continuous DNA synthesis on H and O strand. The replication initiation begins from OH Strand, which proceeds unidirectional. During OL replication stem loop structure is formed which block mtSSB from binding, initiating primer synthesis. Thus the two strand synthesis occur in a continuous manner , until two complete double stranded DNA is formed.A triple-stranded displacement loop structure also known as D Loop is formed, When 7S DNA remains bound to parental L strand, while parental H-Strand is displaced. The role of mtDNA D loop is not completely understood.
Mitochondrial Diseases:
A dysfunction in mitochondria leads to mitochondrial disorder. Heteroplasmy is condition due to presence of mutant mitochondrial DNA . Various Mitochondrial disorders are as follows:
Mitochondrial Myopathy: Presence of ragged red muscle fibres is due to accumulation glycogen and neutral lipids which leads to decreased reactivity of cytochrome c oxidase
Leber’s hereditary optic neuropathy : This is maternally inherited, which causes degeneration of retinal ganglion cells
Leigh syndrome: It is a neurometabolic disorder affecting CNS( central nervous system)
Myoneurogenic gastrointestinal encephalopathy : Autosomal recessive disorder. It is due to mutation of TYMP gene
Mitochondrial DNA depletion syndrome: It is also known as Aplers disease. This is caused my mutation inTK2 gene.
Chromatography, broadly, is a technique to separate two phases which are mutually immiscible. The phases are brought in contact with each other and one of these phases is stationary while the other phase is a mobile phase which moves over the surface of the stationary phase or percolates it. The interactions of the sample mixture which are established after the movement of the mobile phase over the stationary phase lead to the separation of the desired compound present in the mobile phase based on the differences in the physio-chemical properties of the two mediums.
There are various techniques used in the process of chromatography like plane chromatography, paper chromatography, thin layer chromatography, column chromatography etc.
Based on these techniques, further different types of chromatographic procedures have been curated which include adsorption chromatography, partition chromatography, gel permeation chromatography, ion exchange chromatography and affinity chromatography.
GEL PERMEATION CHROMATOGRAPHY:
Gel permeation chromatography is a chromatographic procedure that separates molecules based on their molecular size. This method has various other names such as molecular sieve, gel filtration and molecular exclusion chromatography.
This method is widely used due to its many advantages, some of which are:
It is a very gentle technique that permits the separation of labile molecules.
It is a technique in which the solute recovery is almost 100%.
The reproducibility of this technique is high.
The technique is not very time consuming and is relatively inexpensive.
PRINCIPLE:
The principle of gel permeation chromatography is relatively very simple.
A long column filled with gel beads or porous glass granules is allowed to attain equilibrium with a solvent which is suitable for the separation of the desired compound or molecule.
Assuming that the mixture to be separated contains molecules of varying sizes, when this mixture will be passed through the column containing the beads;
The larger molecules pass through the interstitial spaces between the beads and do not enter the spaces inside the beads. This occurs because the diameter of the molecule is larger than the pore size of the beads.
Therefore, the larger molecules are able to pass down the column with little or no resistance.
The small molecules however, have a size which is smaller than the diameter of the pores of the beads and therefore these molecules enter the beads and reach the end of the column after a longer period of time due to the resistance that is created by the passage of the molecules through the beads.
The degree of retardation (or the extent of the time taken by the molecule to reach the bottom) is proportional to the time it spends inside the pores of the gel which is a function of the molecules pore size and the molecules size.
The molecules which have a diameter equal to or smaller than the pore size, do not enter and are said to be excluded. Therefore, the exclusion limit of a gel can be defined as the molecular weight of the smallest molecule which is not capable of entering the pores. For e.g. linear polysaccharides and fibrous proteins have a much lower exclusion limit as compared to the globular proteins of comparable molecular weight.
TYPES OF GELS USED:
A good gel filtration medium should possess a few properties or characteristics like:
The material of the gel should be chemically inert.
It should contain a small number of ionic groups.
The material of the gel should have a wide variety of particle and pore sizes.
It should have a uniform particle and pore size.
The material of the gel should be of high mechanical rigidity.
Some examples of the types of gels used in gel permeation chromatography include sephadex, polyacrylamide, agarose, styragel etc.
TECHNIQUE/PROCESS OF GEL PERMEATION CHROMATOGRAPHY:
This chromatographic technique can be performed wither by column or by thin layer chromatography techniques.
The initial step of the process is:
Preparation of the beads:
Prior to use, the gels used in the column must be converted to their swollen form by either soaking them in water or using a weak salt solution. The greater is the porosity of the gel beads, more will be the time taken for them to attain equilibrium and reach their maximum size.
If porous glass granules are used, the gel beads need not be hydrated at all.
Preparation of the column:
The gel beds, in their swollen form, are mounted or supported on a column on a glass wool plug or nylon net and the previously swollen gel is added in the form of slurry. The preparation is then allowed to settle. Air bubbles must be removed by connecting the column to a vacuum pump. It must be noted that the level of the liquid must not go lower than the top of the bed.
Addition of the sample:
The sample must be added from the top of the column using a funnel and the volume of the sample that must be added depends on the size and the type of the gel that is used in the preparation of the stationary phase.
Collection of the sample:
The eluant used is steadily added and the effluent can be collected in various various fractions in separate tubes.
Detection of sample:
The most common detection methods include the collection and analysis of the fraction and continuous methods in which the UV absorption, refractive index or radioactivity is measured.
APPLICATIONS:
Gel permeation chromatography is mainly and widely used for the separation of biomolecules and purification. Biomolecules like proteins, hormones, enzymes etc. can be separated using this technique.
This technique is especially useful for the separation of 4S and 5S tRNA.
This technique is used for the separation of salts and small molecules from macromolecules.
Gel permeation chromatography is also chiefly used for the detection of molecular weights of macromolecules.
Sources:
Biophysical Chemistry Principles and Techniques by Updhyay and Nath
Chromatography is a separation and purification technique which is used for the separation of solutes in a mixture, biomolecules etc. on the basis of distribution of the sample to be separated between stationary phase (phase which is not mobile and is usually mounted against a support like a chromatographic column) and the mobile phase (which is continuous/is poured or passed over the stationary phase)
Ion Exchange Chromatography is a method used for the separation as well as purification of ionically charged biomolecules like proteins, polynucleotides, nucleic acids etc.
This technique finds a wide array of applications in the scientific world because of its simplicity and high resolution.
PRINCIPLE:
The process of ion exchange can be defined as the reversible exchange of the ions present in a solution, with the ions electrostatically bound to the inert support medium.
The main factor which governs the process of ion exchange chromatography is the electrostatic force of attraction present between the ions. This electrostatic force of the ions depends on their relative charge, radius of the hydrated ions and the degree of non-bonding
interactions.
Usually, ion exchange separations are carried out in columns packed with an ion-exchanger. The ion-exchanger is a support medium which is inert and insoluble. The medium may be capable of covalently binding to positive (anion exchanger) or negative (cation exchanger) functional groups. The ions which do bind to the exchanger electrostatically are called counterions.
The conditions of separation can be manipulated in such a way that some compounds are electrostatically bound to the ion exchanger while some are not, therefore, helping in the separation of the desired compound.
The sample which contains the sample to be separated is allowed to percolate through the exchanger for a certain amount of time that will be sufficient for the equilibrium of the ions to be achieved.
E– Y– E– X+ + Y+
In the equation mentioned above,
E– : Charged cation exchanger.
Y+ : Counterion of the opposite charge associated with the exchanger matrix.
X+ : Charged molecule bearing a charge similar to the counterion to be separated. This molecule is capable of exchanging sites with the counterion as shown above.
Once the exchange of the counterion with the sample has been achieved, the rest of the uncharged and like charged species is washed out of the column.
The ions that did bind can then be eluted out by either percolating the medium with increasing concentration of Y+ (works by increasing the possibility that the Y+ will replace the X+ in the above-mentioned equation due to it being present in a higher concentration). The elution can also be carried out by increasing the pH of the solvent and hence converting X+ to an uncharged species.
Simply put, once the sample containing the specific ions to be separated it passed through the ion exchanger column, the sample ions (which act as counterions to the ions of the exchanger column) bind with the ions on the exchanger column and form associations. However, the ions of the same charge as the exchanger column, present in the sample solution, repel each other and, therefore, do not bind and pass through the column.
The principles which have been mentioned above also apply to other macromolecules such as proteins and nucleic acids which are capable of showing the presence of both positive and negative ions. The type of molecules can bind to both anionic and cationic exchangers.
TYPES OF ION EXCHANGE RESINS:
The two main types of materials used to prepare ion exchange resins are:
Polystyrene, and
Cellulose
Polystyrene resins are prepared by the polymerization reaction of styrene and divinyl benzene. These resins are very useful for separating compounds with a small molecular weight.
Cellulose based resins have a much greater permeability to macromolecular polyelectrolytes as compared to polystyrene resins and they also possess a much lowers charge density.
Based on the type of charge carried by these ion exchangers and their strength, the ion exchange resins can broadly be classified into four types:
Strong cationic exchange resins:
Weak cationic exchange resins:
Sulphonated polystyreneSulphopropyl cellulose
Condensed acrylic acidCarboxymethylcellulose
Strong anionic exchange resins:
Weak anionic exchange resins:
Polystyrene with -CH2NMe3ClDiethyl (2 hydroxypropyl)quaternary amino cellulose
The buffer is that component of the chromatographic column that helps in the maintenance of the pH. The choice of these buffers is usually dictated by the compounds to be separated and whether the ion exchange is anionic or cationic.
Anion exchange chromatography should be carried out with cationic buffers.
Similarly, cation exchange chromatography should mostly be carried out with only anionic buffers for satisfactory separation and results.
Examples of some buffers used in this technique are:
Ammonium acetate
Ammonium formate
Pyridinium formate
Ammonium carbonate etc.
APPLICATIONS OF ION EXCHANGE CHROMATOGRAPHY:
The most significant application of ion exchange chromatography is in amino acid analysis.
This technique is used to determine the base composition of nucleic acids.
Ion exchange chromatography is used as a method of purification of water. Water is completely deionized using this technique.
This technique is used for the ultra-purification of metal ion free reagents.
It can also be used for the separation of a varied number of vitamins, biological amines, organic acids as well as bases.
Source:
Biophysical Chemistry principles and techniques – Upadhyay and Nath
A brief overview of the infections of the respiratory tract and its pathogens:
The respiratory tract along with the gastrointestinal tracts is one of the major connections between the interiors of the body and the outside environment.
The respiratory tract is the pathway is that pathway of the body through which fresh oxygen enters the body and removes the excess carbon dioxide which is not needed by the body.
Anatomy of the respiratory system:
Broadly, the respiratory system of humans can broadly be divided into two distinct areas; the upper and the lower respiratory tracts.
The parts that consist the lower respiratory tract are:
Trachea
Bronchi, and
Bronchioles
The respiratory pathway begins with the nasal and the oral passages. These passages serve to humidify the air that is inspired. These pathways extend past the nasopharynx and the oropharynx to the trachea and then to the lungs.
The trachea is the organ that divides into the bronchi, which then further subdivides into the bronchioles. The bronchioles are the smallest branches of the trachea which finally terminate into the alveoli.
Approximately 300 million alveoli are said to present in the lungs. These mainly serve as the primary, microscopic, gas exchange structures of the respiratory tract.
The lungs (along with the respiratory system) and the heart lie in the thoracic cavity.
The thoracic cavity has three partitions that are separated from one other by the pleura (the pleura majorly cushions the lungs and reduce the friction which may develop between the lungs, rib cage and the chest cavity. It is a two layered membrane covering the lungs.)
The lungs occupy the right and the left pleural cavity while the mediastinum (the space between the right and the left lungs) is occupied by the esophagus, trachea, large blood vessels along with the heart.
Pathogenesis of the respiratory tract:
The success of an organism to cause disease is mainly dependent on the organism’s ability to cause disease (pathogenesis), and
The human hosts ability to prevent the infection (strength of the host’s immune system)
The host factors that help in non-specifically protect the respiratory tract from infection are:
Nasal hair
Convoluted passages and the mucous lining of the nasal turbinate
Secretory IgA and non-specific antibacterial substances (like lysozyme) in respiratory secretions
The cilia and the mucous lining of the trachea and reflexes such as coughing and sneezing.
In addition to the non-specific hosts defenses, normal flora of the nasopharynx and the oropharynx help in the prevention of colonization of the upper respiratory tract.
Microorganism factors:
Organisms possess certain traits that promote colonization leading to infection in the host. The factors that influence the respiratory tract infections are –
Adherence:
The potential of a microorganism depends, in one way or the other, on its ability to establish a stable contact/foothold on the surface of the host by the process of adherence.
The ability of microorganisms to adhere to the host surface is dependent on two factors:
Presence of normal flora, and
Overall state of the host.
Surviving or growing on host tissue without causing harmful effects is called colonization.
Most etiologic agents must first adhere to the mucosa of the respiratory tract to some extent before they can cause harm.
Example: Streptococcus pyogenes possess specific adherence factors and its gram-positive cell wall contains lipoteichoic acids and M proteins. Many gram-negative bacteria like Enterobacteriaceae, Pseudomonas spp., Bordetella pertussis, adhere by the means of proteinaceous fingerlike projections called fimbriae.
Viruses possess either a hemagglutinin or other proteins that mediate that epithelial attachment.
Toxins:
Certain microorganisms are considered to be etiologic agents of disease because they possess virulence factors that are expressed in every host.
Example: Corynebacterium diphtheriae.
Some strains of Pseudomonas aeruginosa also produce toxins which are similar to the toxins of Diphtheria.
Bordetella pertussis which is the causative agent of whooping cough produces toxins that play a role in inhibiting the activity of phagocytic cells and damaging the cells of the respiratory tract.
Microorganism growth:
Pathogens cause disease by merely growing in the host tissue, interfering with normal tissue function and attracting host immune effectors, such as neutrophils and macrophages.
Example: S. pyogenes, M. tuberculosis, Mycoplasma pneumoniae, etc.
Avoiding the Host Response:
Certain respiratory tract pathogens possess the ability to evade host defense mechanisms.
S. pneumoniae, H. influenza, K. pneumoniae and others possess polysaccharide capsules that serve both to prevent engulfment by phagocytic host cells and to protect somatic antigens from being exposed to host immunoglobulins.
Organisms of the respiratory tract and agents that cause diseases:
Pathogens may or may not cause the respiratory infection but can be present as a part of normal flora.
Some of the pathogens that exist and results in the respiratory infection are referred to as true pathogens.
Some of the pathogens that are present in the body but never cause an infection until and unless they are met with the favorable conditions are called
as opportunistic pathogens.
Possible pathogen: they are the pathogens that are likely to cause respiratory
infections.
Example: Actinomyces spp., Haemophilus influenzae, Enterobacteriaceae, etc.
Rare pathogen: pathogens that may cause a respiratory infection are rare
pathogens. Example: Coxiella burnetti, Brucella spp., Salmonella spp, etc.
Definite respiratory pathogen: pathogens that always cause respiratory infections are called as definite respiratory pathogens.
Example: Bordetella pertussis, Blastomyces dermatitidis, Legionella spp., etc.
Different types of agents that cause respiratory diseases are bacteria, fungi or
viruses.
Bacterial agents: the bacterial agents that cause respiratory infections are
Mycoplasma spp., Streptococcus pneumoniae and Neisseria meningitides.
Fungal agents: the fungal agents that cause respiratory infections are Candida
albicans, Cryptococcus neoformans and Histoplasma capsulatum.
Viral agents: the viral agents that cause respiratory infections are human
metapneumovirus, adenovirus, enteroviruses, and herpes simplex virus.
Major respiratory diseases are caused by M. tuberculosis, S. pyogenes and
K.pneumonia.
Sources:
Foundations in Microbiology, Author: Kathleen Park Talaro, Barry Chess
A hormone is a signaling molecule secreted by endocrine glands in response to physiological stimuli in multi cellular organisms. These hormones circulate in blood and reach its destination to exert specific function. Hormones help maintain physiological and behavioral functions in the organisms.
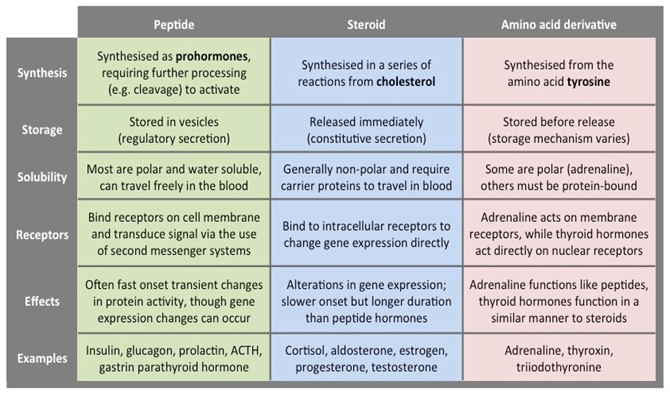
Hormones are classified into three main classes:
Steriod hormones – Lipid soluble and move across plasma membrane of the targeted cells.
Peptide hormones – Water soluble and act through cell surface receptors present on the targeted cells.
Aminoacid derivatives
Table 1: Three classes of hormones
Peptide hormones
Peptide hormones are made up of small amino acid chains called, peptides. Peptide hormones are synthesized in the cells from amino acids based on mRNA sequence that is derived from DNA template within the nucleus.
Peptide hormones can’t navigate across plasma membrane of the cell. Hence, they exert their function by binding to the receptor present on the cell surface of the target cell that in turn trigger signal transduction and cellular response. Some peptide hormones like parathyroid hormone-related protein, angiotensin II etc interact with intracellular components within cytoplasm or nucleus by an intracrine mode of interaction.
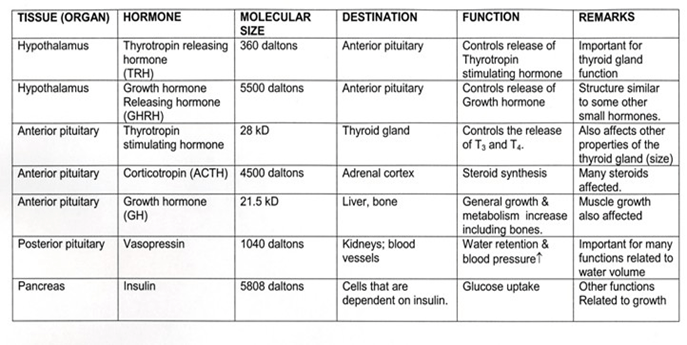
Some of the examples of peptide hormones are as follows:
Adrenocorticotrophic hormone
Thyroid stimulating hormone
Vasopressin
Angiotensin II
Antrial natriuretic peptide
Calcitonin
Follicle stimulating hormone
Insulin
Growth hormone
Parathyroid hormone
Prolactin
Table 2: Main peptide hormones – details and functions
Detection of peptide hormones
As peptide hormones circulate in blood to reach their destination, serum generally acts as the source of detection and measurement.
The following are the main detection methods used to detection peptide hormones:
Sandwich ELISA technique:
In sandwich ELISA method, two antibodies are used to detect hormone of interest. One of the antibodies is attached to the solid support on the micro titer plate, called as capture antibody.
Second antibody labeled with a signal molecule (enzyme or radioisotope or chemilumiscent) acts as detector.
When the analyte containing mixture is loaded onto the micro titer plate, capture antibodies bind to the analyte via epitope that is present on the analyte surface and catch hold of analytes.
When the detector antibodies are added to the plate, they bind to different location on the analyte.
In the enzyme based reaction, when substrate is added, it reacts with enzyme that is attached to the detector body and shows response in the form of color change.
Figure 1: Sandwich ELISA to detect analytes in the blood
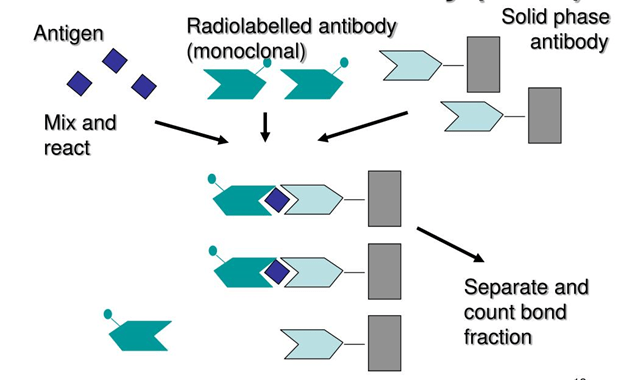
Radioimmunoassay (RIA):
RIA is an in vitro detection technique that detects and measures antigens (like hormones and other foreign substances) in the blood. RIA technique is discovered by Berson and Yalow in 1960 to analyze insulin levels in blood.
RIA method is based on the radioactivity measurement associated with antigen-antibody interactions in the reaction.
A known antigen that is radiolabelled is incubated with antibody at known concentrations.
When the analyte containing solution is added to the labelled antigen-antibody mixture, antigen of interest replaces labelled antigen and bind to the antibody.
More the antigen of interest present in the solution, more labelled antigen will be displaced and replaced with antigen of interest.
Figure 2: Radioimmunoassay
Enzyme multiplied Immunoassay Technique (EMIT):
EMIT is more easy and equivalent detection method, both qualitatively and quantitatively to measure wide-range of analytes from the serum.
EMIT is based on the principle that the amount of analyte present in the solution is directly proportional to the inhibition of enzyme-substrate reaction complex.
In this technique, initially a known analyte is labelled with an enzyme and antibody specific to drug is allowed to bind drug-enzyme complex. This results in inhibition of enzyme activity.
When the solution containing analyte is added to the above mixture, the analyte releases the antibody from the drug-enzyme complex, thereby increasing enzymatic activity.
Therefore, enzyme activity is proportional to the analyte present in the sample added and is measured by absorbance value changes of the enzyme.
Figure 3: Enzyme multiplied Immunoassay Technique
Immunoradiometric assay (IRMA)
IRMA utilizes radiolabeled antibodies to detect analytes of interest.
In this technique, antibody is directly labeled with radioisotopes rather than using two antibodies as in other immune assays.
When an analyte containing solution is added to micro titer plateradiolabeled antibodies bind to the specific epitopes of the anlytes and forms the antigen-antibody complex.
I125 and I131 radioisotopes used in general for this assay.
Unbound radiolabeled antibodies are removed from the plate by second wash during the process.
Figure 4: Immunoradiometric assay
The other peptide hormone detection methods are listed below:
In 1952, Joshua Lederberg invented the word ‘plasmid.’ It was initially formed from bacteria; plasmids are extrachromosomal genetic elements that can reproduce independently in most Archae, Eukarya species, and Eubacteria. Plasmids are double-stranded circular DNA molecules that are different from the chromosomal DNA of the cells.
The genetic material found within the chromosomal DNA guides the structure and function of a bacterial cell. In some instances, plasmids are usually not required for the host bacterium to survive. Although not necessary, by encoding functions that may not be defined by the bacterial chromosomal DNA, plasmids significantly contribute to bacterial genetic diversity and plasticity. Antibiotic tolerance and the expression of proteins, for example. The plasmid is also encoded by antibiotic resistance genes, allowing the bacteria to survive in an environment containing antibiotics, thereby providing the bacterium with a competitive advantage over species susceptible to antibiotics. Plasmids may be altered as a means to express the protein of interest for e.g., production of human insulin using recombinant DNA technology.
As a mechanism for gene-cloning and as a vehicle for gene-expression, plasmids have been central to modern recombinant DNA technology.
Isolation of Plasmids
In molecular biology, bacterial plasmid DNA isolation is a crucial technique and an integral step in many processes, such as cloning, DNA sequencing, transfection, and gene therapy. These manipulations need the isolation of high purity plasmid DNA. In all molecular biology procedures, cloning, such as digestion with restriction enzymes, PCR, transfection, in vitro translation, blotting, and sequencing, purified plasmid DNA can be used for immediate usage.
In molecular biology, alkaline lysis is used to separate plasmid DNA or other cell components, such as proteins, by splitting open the cells. Primarly, bacteria containing the plasmid of interest are allowed to grown and then lysed with an alkaline lysis buffer composed of a sodium dodecyl sulphate (SDS) detergent. Cellular debris is extracted, and the plasmid is separated and filtered through a series of steps, including agitation, precipitation, centrifugation, and the removal of the supernatant.
Principle:
Purification of plasmid DNA from bacterial DNA using alkaline lysis is based on the differential denaturation of plasmid and chromosomal DNA. Disruption of the cellular structure to produce a lysate, separation of the plasmid from the chromosomal DNA, cell debris, and other insoluble material are the essential steps of plasmid isolation. With a lysis buffer solution, bacteria are lysed.
Materials and Equipment:
Refrigerated centrifuge
Vortex
Microwave oven
pH meter
Orbital shaker
Micropipettes
Autoclave
LB plate with Bacterial colonies
1.5 ml micro-centrifuge tubes
Autoclaved distilled water
Microtips
Microfuge tubes
Chemicals:
1. Lysis Solution (Solution I)
2. Denaturation solution (Solution II)
3. Neutralizing solution (Solution III)
4. TE Buffer
5. RNase
6. Phenol: Chloroform: isoamyl alcohol
7. 70% Ethanol
8. Isopropanol
Preparation of Stock solutions:
Solution I (Lysis solution): 50mM Glucose, 25mM Tris-Hcl (pH 8.0), 10mM EDTA (pH 8.0). Store at 4oC.
Solution II (Denaturation solution):1% SDS, 0.2N NaOH (pH 12.0). Freshly prepared and store at room temperature.
Solution III (Neutralizing solution): 60 ml of 5 M potassium acetate, 11.5 ml of glacial acetic acid, and 28.5 ml of water. Store at 4oC.
TE Buffer: 10mM Tris-HCl (pH 8.0), 10mM EDTA (pH 8.0)
RNase (1mg/ml)
Phenol: Chloroform: Isoamyl alcohol -(25:24:1).
Washing buffer
Elution buffer
Biological material:
Overnight grown culture of E.coli.
PROCEDURE
Harvesting of the cells:
The single bacterial colony is transferred into a 5 ml LB medium containing appropriate antibiotic, incubated for 16 hrs at 37oC with vigorous shaking.
About 1.5 ml culture is transferred into a microfuge tube and centrifuged at 6000 rpm for 5 min at 4oC and discard the supernatant.
Isolation of Plasmid by alkyl-lysis method:
1. The bacterial pellet is re-suspended in 100μl of ice-cold solution-I by vigorous vortexing. This is essential to ensure that the bacterial pellet is wholly dispersed in the solution I.
2. Add 200μl of freshly prepared solution II. Mix the contents by inverting the tubes rapidly 4 to 5 times. A transparent and viscous solution ensured complete lysis. Do not vortex. Incubate on ice for 3 min.
3. Add 150μl of ice-cold solution III. Mix by gently inversion for about 10 seconds to disperse the solution III through the vicious bacterial lysate. Further incubate the tubes on ice for 5 min.
4. Centrifuge at10,000 rpm for 5 minutes at 4oC and transfer the supernatant to a fresh tube.
5. To this add equal volumes of phenol-chloroform, mix by vortexing. After centrifuging at 10,000 rpm 5 minutes at 4oC, transfer the aqueous phase to a fresh tube.
6. Precipitate the double-stranded DNA by adding an equal volume of isopropanol, mix gently, and allow the mixture to stand for two minutes. Then centrifuge at 10,000 rpm for 10 minutes at 4oC.
7. Remove the supernatant by gentle aspiration.
8. Then wash the pellet with 1 mL of 70% ethanol (twice). Allow the DNA pellet to air dry for 10 minutes.
9. Re-dissolve the DNA pellet in 50μl of TE (pH 8.0).
Isolation of PLASMID by spin column method:
1. Inoculate a single colony of bacterial cells from the petri plate using a sterile inoculation loop to 5ML of LB broth and allow them to grow overnight at 37°C at 200 rpm.
2. From the pre-inoculum transfer, 1.5 ML of bacteria-containing cells into 2mL centrifuge tubes and centrifuge the cells at 8000 rpm. for one minute. Then the supernatant was discarded.
3. Re-suspend the cells in 200 µL of resuspension solution. Then mix the cells thoroughly by pipetting up and down or vortexing it.
4. To this, add 200 µL of lysis solution and mix the tubes by gently inverting up and down.do not vortex it. They were then incubated for five minutes.
5. To this, add 350 µL of the neutralizing solution and, for mixing, invert the tubes 4 to 6 times. Then Centrifuge these tubes at 10,000 rpm for 10 minutes.
6. Binding column preparation: Take a new centrifuge tube and insert a new binding or spin column into it. To this, add 500 µL of column preparation solution and spin at 10,000 rpm for one minute. Then discard the solution that was collected in the centrifuge tube.
7. Then, transfer the clear lysate to the binding column and centrifuge at 10,000 rpm for one minute. Discard the flow-through that was remained in the centrifuge tube.
8. Washing: In this step, add 750 µL of washing solution to the binding column and spin the centrifuge tube for one minute. Then discard the solution that remained in the centrifuge tube. (Do the washing step twice).
9. Then, change the spin column into the new centrifuge tube. Then spin the column for one minute and incubate at room temperature for 2 – 5 minutes.
10. ELUTION: In this step, the spin column is transferred to the fresh collecting tube. Then add 70 µL of elution buffer to the spin column and spin for one minute at 10,000 rpm. Label the vial as elution I.
11. Transfer the same spin column to the fresh collecting tube and add 40 µL of elution buffer to the spin column and spin for one minute at 10,000 rpm. Label the vial as elution II.
12. Then discard the spin column, and the vials labelled as elution I and II are stored at -20oC or -80oC.
Preparation of agarose gel electrophoresis:
First, take a gel casting tray, then clean the tray with ethanol and seal the edges with transparent tape and place the comb.
Then 0.8% of agarose solution was prepared using 1X TBE. When the temperature was lowered, add a little amount of Ethidium bromide to it and mix well.
After mixing, pour the agarose solution into the gel casting trays and then allow for polymerization. Make sure with the absence of air bubbles while pouring the solution. After polymerization, remove the comb without breaking the gel.
Submerge the agarose gel into the electrophoresis tank containing 1X TBE buffer.
Electrophoresis of isolated plasmid DNA.
1. Take 3 – 4 µL of eluted sample and add 2 µL of sample buffer.
2. Load 5-6 µL of a sample into each well along with a DNA marker or ladder.
3. Run the electrophoresis and allow the plasmid DNA to run in 1X TBE buffer at a constant voltage. Keep track of the dye front.
4. Then disconnect the power electrophoresis tank and remove the gel using gloves. Place the gel in the Gel Documentation System or Transilluminator and visualize the DNA bands in the UV light.
5. Quantify the concentration of DNA from the DNA marker or ladder used.
Role of chemicals used:
Spin column: The desired nucleic acids should be bound to the column after centrifuging the lysate through the silica membrane, and impurities such as protein and polysaccharides should be in the flow-through. While plant samples are likely to contain polysaccharides and pigments, the membrane may be slightly brown or yellow in blood samples. The washing steps will remove such impurities. Usually, there are two washing measures, but this varies depending on the type of sample. A low concentration of chaotropic salts to eliminate residual proteins and pigments will also be part of the first wash. In order to extract the salts, this is often accompanied by an ethanol wash. Columns contain a resin of silica which binds to DNA/RNA selectively. By its capability to bind silica in the presence of high concentrations of chaotropic salts, the DNA of interest can be isolated.
Washing: With an alcohol-based wash, these salts are then removed, and the DNA is eluted using a low-ionic-strength solution such as TE buffer or water. Dehydration and the formation of hydrogen bonds that compete against poor electrostatic repulsion seem to be driving the binding of DNA to silica. Therefore, a high salt concentration will help push DNA adsorption to silica, and the DNA will be released at a low concentration.
Elution buffer: The Elution buffer is initially used to wash away unbound proteins and release the ligand’s desired protein at a higher concentration. The elution buffer must work rapidly without altering the desired protein’s function or activity. This enables to stand within the membrane for a few minutes before centrifugation for optimum DNA elution.
Glucose: It helps in maintaining osmolarity and keeps the cells from bursting.
Tris HCL and EDTA: This helps in chelating divalent metal ions such as magnesium calcium and destabilizes the cell wall, inhibiting DNases’ action.
NaOH and SDS: In the lysis buffer or solution II, sodium hydroxide, and the detergent Sodium Dodecyl (lauryl) Sulfate (SDS) are present. SDS is used to solubilize the cell membrane. NaOH helps in the breakdown of the cell wall, but more significantly, it interferes with the hydrogen bonding between the DNA bases, turning the cell’s double-stranded DNA, including the genomic DNA and the plasmid, into single stranded DNA.
RNase: It helps incomplete digestion of unwanted RNA from the plasmid sample.
Galactose is a sweet tasting monosaccharide sugar which is a carbohydrate, which is an aldohexose molecule. Galactose is a very important molecule in the body as it is needed for the synthesis of lactose, glycolipids, glycoproteins as well as proteoglycans. It is also known as “brain sugar” as it is useful in formation of the glycolipids which occur in the brain and the myelin sheaths of the nerve cells. Galactose can be hydrolysed from interstitial disaccharide lactose, which is known as the ‘sugar of milk’. (lactose = glucose + galactose). Glucose and galactose are stereoisomers of each other, differing in stereochemistry at carbon 4.
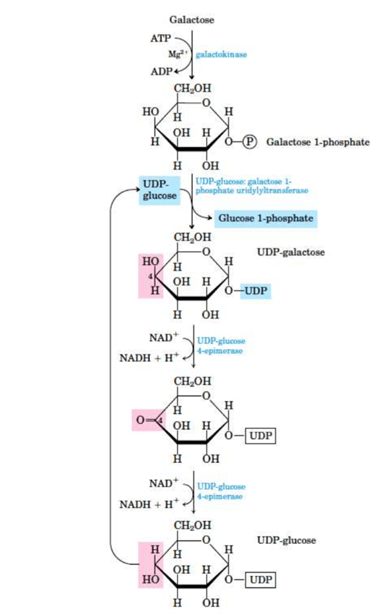
Conversion of galactose to glucose
Galactose can be readily converted to glucose in the liver. This is done in the following steps:
The first step is the conversion of galactose to galactose-1-phosphate on the action of galactokinase. It is a phosphorylation reaction, in which ATP acts as the donor of the phosphate group.
Galactose-1-phosphate reacts with UDP glucose i.e. uridine diphosphate glucose (UDPGlc) to form uridine diphosphate galactose (UDPGla). Glucose-1-phosphate is also formed and given out in this step.
This reaction is catalysed by an enzyme called galactose-1-phosphate uridyl transfer
After this, UDP galactose which is formed is converted to UDP glucose by an epimerization reaction.
This is catalysed by UDP-4 glucose epimerase.
The epimerization reaction will involve the oxidation and reduction of carbon 4, using NAD+ as coenzyme.
Finally, from the so formed UDP glucose, glucose is released in the form of glucose 1 phosphate
NOTE: The epimerization reaction is freely reversible, thus glucose can convert back to galactose as and when required. This is the reason that galactose is not a dietary essential.
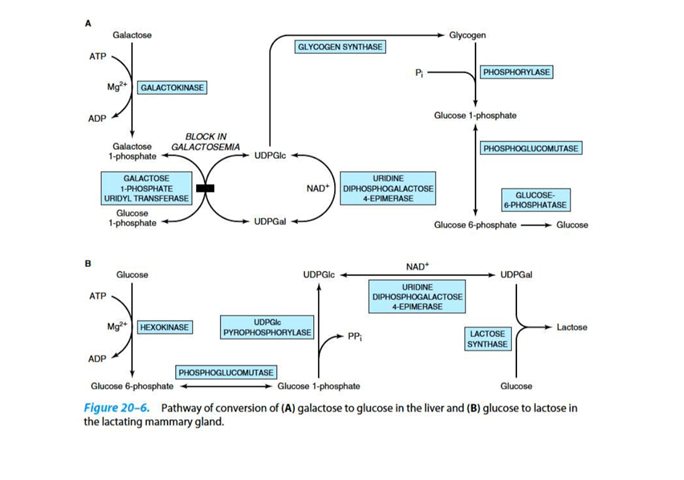
To yield lactose in the mammary gland, UDPGal condenses with glucose. This reaction is catalysed by lactose synthase
Galactosemia is a disease related to this pathway:
Galactosemia is the disruption of galactose metabolism.
Classic galactosemia is the most common type, in which a deficiency of galactose 1 phospahte uridyl transferase activity is inherited.
Due to this, galactose 1 phosphate is accumalated and as a consequence, the inorganic phosphate of the liver is depleted.
This results in the ulitimate liver failure and mental detoriation.
Infants who are afflicted seriously fail to thrive. They go through symptoms of vomitting and diarhhea after consuming dairy products, enlargement of the liver, and jaundice (which can progress to cirrhosis)
Cataract, lethargic developement and retarded mental developement is also very common amongst children. Cataract is observed due to deposition of a galactose metabolite, galactitol, in the lenses of the eyes. Galactitol is osmotically active, due to which water diffuses in the eyes and instigates cataract formation.
Other side effects include increase in level of blood galactose level, which is then found in urine.
Females can also display ovarian failure.
A stirct limitation of galactose consumption in diet can greatly reduce these symptoms.
An important diagnostic criterion for this the absence of transferae in the red blood cells.
Conclusion:
Thus, galactose is a very important carbohydrate present in the body which can be easily converted to glucose ( in the form of glucose-1-phosphate) for energy. This is a reversible reaction, as glucose and galactose are stereoisomers. This is why galactose is not an essential need for the body.
Deficiency in the enzymes that carry out this conversion may result in a vast range of symptoms under galactosemia.
The potential for spreading through drinking water is the emerging pathogenic bacteria of concern outlined here, but they do not correlate with the existence of E. Coli or with other measures of the consistency of drinking water widely used such as coliform bacteria. There are no satisfactory microbiological markers of their existence in most cases. To understand the real nature and dimension of the diseases caused by water polluted with these bacteria and the ecology of these pathogens, further studies are required.
Mycobacterium Avium Complex (Mac):
The complex Mycobacterium avium (Mac) consists of 28 serovars of two species: Mycobacterium avium and Mycobacterium intracellular. With the discovery of disseminated infection in immunocompromised individuals, especially people with HIV and AIDS, the Mac species’ significance was recognized. MAC members are deemed to be opportunistic human pathogens. A wide range of environmental sources, including coastal waters, rivers, lakes, streams, wetlands, springs, soil, piped water supplies, plants, and house dust, have defined Mac species. Mac species have been isolated from the delivery systems of natural water and drinking water in the USA. The ubiquitous existence of Mac organisms stems from their ability under varied conditions to thrive and evolve. Mac species can proliferate at temperatures up to 51°C in water and expand over a broad pH range in natural waters. These mycobacteria are incredibly resistant to the use of chlorine and other chemical disinfectants in drinking water care. Standard drinking-water treatments may not remove Mac species but may substantially reduce the numbers present in the source water to a level that poses a negligible risk to the general public if it is running satisfactorily. In delivery systems, the entryway for these mycobacteria is through leaks. For their continued presence in distribution systems, the growth of Mac organisms in biofilms is probably significant.
Slow-growing mycobacteria can be present in the surface biofilm at densities higher than 4,000 per cm2, producing a potentially high exposure level. The signs of Mac infections result from either respiratory or gastrointestinal colonization, potentially spreading to other places in the body. Exposure to Mac species may occur through the consumption of contaminated foodstuffs, the inhalation of air containing contaminated soil particles, or through touch or ingestion, aspiration or aerosolization of the organisms containing drinking water. Unlike gastrointestinal pathogens, where E. No appropriate indicators have been identified to signal increasing Mac species concentrations in water systems, and coli can suggest possible presence.
Helicobacter pylori:
As a significant etiologic agent for gastritis, Helicobacter pylori has been cited and has been involved in the pathogenesis of duodenal ulcer and peptic disease and gastric carcinoma. Most people who are infected by this pathogen, however, remain asymptomatic. Using methods based on history, H. There has been no isolation of pylori from environmental sources, including water. Molecular methods have, on the other hand, been useful in detecting the pathogen.
Fluorescence in situ hybridization (FISH) has been successfully used to detect this pathogen in drinking water delivery systems and other water bodies. To detect the presence of H, a polymerase chain reaction was also used. Pylori DNA in drinking water, especially biofilm-associated. In biofilms for drinking-water, H. Pylori cells lose culturability rapidly and enter a viable but non-culturable state. Cells persist for more than one month in these biofilms, with densities exceeding 106 cells per square cm. It remains unclear how the organism is transmitted. Nevertheless, the fact that it has been oral-oral or fecal-oral transmission is demonstrated by recuperation from saliva, dental plaques, stomach, and fecal samples. Water and food tend to be of less immediate significance, but they can still play a significant role in improper sanitation and hygiene.
Aeromonas Hydrophyla:
Over the past years, A. Hydrophila has received attention as an opportunistic pathogen for public health. The elderly, children under the age of five, and immunosuppressed persons may play a significant role in intestinal disorders. Gram-negative, non-spore-forming, rod-shaped, facultative anaerobic bacilli belonging to the Aeromonadaceae family are Aeromonas hydrophila. Even though the dominant species is typically hydrophila, whereas other aeromonads, such as A.Sobria, and A.Caviae were isolated from human feces and water sources. Species of Aeromonas, including A. Hydrophila, in the field, are ubiquitous. It is also segregated from food, potable water, and aquatic ecosystems. Concentrations of Aeromonas spp. in safe rivers and lakes Typically, 102 colony-forming units (CFU)/mL are around. In general, groundwater contains less than 1 CFU/mL. It was noticed that drinking water immediately leaving the treatment plant contained between 0 and 102 CFU/mL. Drinking water can show higher concentrations of Aeromonas in delivery systems due to the growth of biofilms. With Aeromonas spp. growth was observed between 5° – 45° C.
A. Hydrophila is immune to standard treatments with chlorine and is likely to live within biofilms. Ingestion of infected water or food or touch of the organism with a break in the skin are the typical routes of infection suggested for Aeromonas. A potential source of pollution for human beings may be drinking or natural mineral water. There was no recorded person-to-person transmission.
Any natural or industrial process that allows free nitrogen (N2), a relatively inert gas abundant in the air, chemically combines with other components to form more reactive nitrogen compounds, such as ammonia, nitrates, or nitrites.
Nitrogen does not react with other elements under ordinary conditions. However, in all fertile soils, in all living organisms, in many foodstuffs, in coal, and such naturally occurring chemicals as sodium nitrate and ammonia, nitrogenous compounds are contained. As DNA, nitrogen is also present in each living cell’s nucleus.
Role of nitrogen in nature:
The growth of all organisms is dependent on the availability of mineral nutrients, and none is more crucial than nitrogen, which, as an integral component of proteins, nucleic acids, and other cellular constituents, is required in large quantities. In the earth’s atmosphere, there is an ample supply of nitrogen – approximately 79 percent in the form of N2 gas. However, since there is a triple bond between the two nitrogen atoms, N2 is unavailable for most species, rendering the molecule virtually inert. It must be ‘fixed’ (combined) in the form of ammonium (NH4) or nitrate (NO3) ions for nitrogen to be used for growth. The weathering of rocks releases these ions so slowly that fixed nitrogen availability has a marginal effect. Therefore, nitrogen is always the limiting factor for the growth and development of biomass in all habitats where there are an adequate climate and water availability to sustain life.
In nearly all aspects of the availability of nitrogen and thus for life support on earth, microorganisms play a central role:
Some bacteria can turn N2 into ammonia; they are either free-living or in symbiotic relationships with plants or other species (e.g., termites, protozoa). Other bacteria cause ammonia transformations to nitrate, and many bacteria and fungi degrade organic matter from nitrate to N2 or other nitrogen gases, releasing fixed nitrogen for reuse. These processes also lead to the cycle of nitrogen.
Examples of nitrogen fixing bacteria
Biological Nitrogen Fixation Process
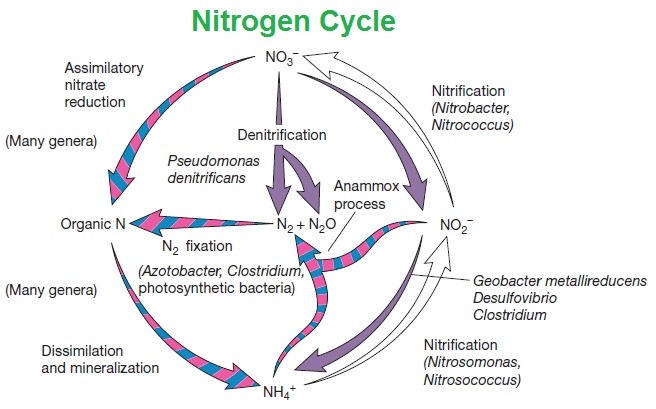
Nitrogen cycle: The cycle of nitrogen is a repeating cycle process in which nitrogen travels through the soil, atmosphere, water, plants, animals, and bacteria, both living and non-living things. Nitrogen must change types in order to pass through the various parts of the cycle. Nitrogen occurs as a gas (N2) in the atmosphere, but it exists as nitrogen oxide, NO and nitrogen dioxide, NO2, in the soils and can be present in other forms when used as a fertilizer, such as ammonia, NH3, which can be further converted into another fertilizer, ammonium nitrate or NH4NO3.
The nitrogen cycle occurs in five steps:
Nitrogen fixation,
Mineralization,
Nitrification,
Immobilization, and
Denitrification.
Microbes in the soil convert nitrogen gas (N2) into volatile ammonia (NH3) in this picture, so the volatilization is called the fixation process. Leaching is when specific nitrogen sources (such as nitrate or NO3) are dissolved in water, escaping from the soil and potentially polluting waterways.
A. NITROGEN FIXATION:
In this process, nitrogen moves into the soil from the atmosphere. A massive reservoir of nitrogen gas is in the earth’s atmosphere (N2). However, this nitrogen is not ‘available to plants because without transforming, the gaseous form cannot be used directly by plants. N2 must be converted by a mechanism called nitrogen fixation in order to be used by plants.
1. In the atmosphere, fixation transforms nitrogen into which plants can absorb through their root systems.
2. When lightning gives the energy required for N2 to react with oxygen, creating nitrogen oxide, NO and nitrogen dioxide, NO2, a small amount of nitrogen can be fixed via rain or snow; these sources of nitrogen then enter the soil.
3. By the industrial process that produces fertilizer, nitrogen may also be fixed.
4. This method of fixation takes place under high pressure and heat, during which nitrogen(atmospheric) and hydrogen, combined to form ammonia (NH3); which can then be further processed for the development of ammonium nitrate (NH4NO3), a form of nitrogen which can be applied to the soil and used by plants.
5. The majority of nitrogen fixation occurs naturally by bacteria in the soil.
6. Some bacteria bind to plant roots and have a relationship with the symbiotic plant.
7. Through photosynthesis, the bacteria get energy, and in exchange, they fix nitrogen in a form that requires the plant. The fixed nitrogen is then transferred to other parts of the plant and used to shape the plant’s tissues to expand.
8. Other bacteria live freely in soil or water, without this symbiotic relationship, and can fix nitrogen. These bacteria can also produce sources of nitrogen that species can use.
B. MINERALIZATION:
This process occurs in the soil from organic sources, such as manure or plant materials, nitrogen transfers to an inorganic source of nitrogen used by plants. The plant’s nutrients are gradually used up, and the plant dies and decomposes. In this stage of the nitrogen cycle, this becomes true.
1. Mineralization occurs when bacteria, such as animal manure or decomposing plant or animal waste, operate on organic material, and begin to transform it into a nitrogen source that plants can use.
2. All plants under cultivation, except legumes, obtain the nitrogen they need from the soil.
3. Legumes get nitrogen through fixation that happens in their root nodules.
4. NH3 is ammonia, the first source of nitrogen formed by the mineralization process. The NH3 in the soil then reacts to form ammonium, NH4, with water.
5. This ammonium is kept in the soils and is accessible via the symbiotic nitrogen-fixing relationship mentioned above for use by plants that do not get nitrogen.
C. NITRIFYING
1. The third step, nitrification, also takes place in the soil. The ammonia formed during mineralization in the soils is converted into nitrites, NO2- and NO3-nitrates during nitrification.
2. The plants and animals consuming the plants will use nitrates.
3. In the soil, some bacteria can convert ammonia into nitrites.
4. While nitrite is not usable by plants and animals directly, other bacteria may convert nitrites into nitrates, a form that is usable by plants and animals.
5. For the bacteria involved in this process, this reaction provides energy.
Nitrosomonas and Nitrobacter are the bacteria that help in fixing nitrogen. Nitrobacter transforms nitrites into nitrates; Nitrosomonas converts nitrites to ammonia. Only in the presence of oxygen can both forms of bacteria function. For plants, the nitrification process is essential as it creates an additional stash of usable nitrogen that can be consumed by the plants via their root systems.
D. IMMOBILIZATION:
Immobilization, often defined as the reverse of mineralization, is the fourth stage of the nitrogen cycle. Together these two processes regulate the amount of nitrogen in the soil. Microorganisms living in the soil, just like plants, require nitrogen as an energy source.
1. When the residues of decomposing plants do not contain enough nitrogen, these soil microorganisms pull nitrogen from the soil.
2. These nitrogen sources are no longer available to plants when microorganisms take in ammonium (NH4+) and nitrate (NO3−) and can cause nitrogen deficiency or a lack of nitrogen.
3. Therefore, immobilization binds up nitrogen in microorganisms.
4. Immobilization, however, is essential because it helps to regulate and balance the amount of nitrogen in microorganisms in the soils by binding it up or immobilizing the nitrogen.
E. DENITRIFICATION:
1. Nitrogen returns to the air in the fifth stage of the nitrogen cycle when bacteria transform nitrates to atmospheric nitrogen (N2) via denitrification.
2. As the gaseous form of nitrogen travels into the atmosphere, it results in an overall loss of nitrogen from soils.
Not enough nitrogen in the soils makes plants hungry, while too much of a good thing can be harmful: plants and even livestock can be contaminated by excess nitrogen! The contamination of our water supplies by excess nitrogen and other nutrients is a significant concern, as the decomposition of dead algae blooms is suffocating marine life. Farmers and communities need to increase crop absorption of added nutrients and adequately manage the excess of animal manure.
Poxviruses are part of the family Poxviridae and can infect humans and animals alike. Smallpox (variola), vaccinia, cowpox, monkeypox, buffalopox, aracatuba, and cantagalo viruses are orthopoxviruses. Orf virus, pseudo cowpox virus, deer poxvirus, bovine papular stomatitis virus, and sealpox virus are parapoxviruses. Yatapoxviruses include the tanapox virus, which is commonly found in Africa. The human poxvirus, the molluscum contagiosum virus, contains mollusc poxviruses.
Smallpox is unique to humans and molluscum contagiosum. In humans, other viruses cause unusual zoonotic infections. The vaccinia virus used for vaccines will infect humans as well. Molluscum contagiosum is also a human-unique poxvirus. Other human poxvirus infections are caused either by zoonotic exposure to animal poxviruses or scheduled or unintended vaccinia administration. This virus is spread through close contact, often via sexual contact.
Recently, laboratory exposures have been reported that have contributed to infection with vaccinia and tanapox viruses, widely used as vectors for experimental vaccines. Due to transmission to contacts, civilian and military personnel’s smallpox vaccination program resulted in multiple infections.
Incubation Period: Infections with smallpox are caused by inhalational penetration to nasal, oral, or pharyngeal droplets. The incubation period is from 10-14. Viruses of smallpox multiply locally and spread to the nearby lymph nodes. On days 3-4, asymptomatic viremia ensues, spreading to the bone marrow and spleen. On approximately day 8, secondary viremia begins. Generalized signs of fever and a toxic appearance are associated with this secondary viremia. In the dermis blood vessels, the virus in leukocytes then becomes localized. Then the stereotypical smallpox rash emerges.
The replication:
1. Generally, poxviruses replicate only in the cytoplasm and can in cells without a nucleus, unlike most DNA viruses.
2. Either through endocytosis or by a fusion event, the virion enters the cell.
3. For later replication cases, the viral core reaches the cytoplasm and functions as a scaffolding.
4. Besides, many necessary enzymes such as viral transcriptase, transcription factors, capping and methylating enzymes, and a poly(A) polymerase are transported by the virus.
5. Therefore viral DNA transcription is rapidly initiated and about 100 early viral genes, especially enzyme coding genes that are involved in viral DNA replication, are triggered.
6. At the same time as DNA replication, transcription of intermediate and late genes is initiated.
7. In specific cytoplasm areas, virus assembly and immature viruses can be visualized very quickly.
8. The Virions travel to the Golgi complex during maturation, where they are enveloped before being released by budding or cell disruption.
9. Some virions are enveloped, and they may have certain benefits, including cell uptake speed.
Other poxviruses, primarily localized diseases, typically follow the same pattern of evolution. An exception is the outbreak of monkeypox, which progresses to a variola-like clinical syndrome. Monkeypox infections, as in the North American epidemic, can vary from mild infections with few lesions to severe systemic diseases that mimic smallpox. At the inoculation site, the Molluscum contagiosum virus also replicates, but the skin lesions’ character is distinct.
Epidemiology:
Human poxvirus infections are obtained from animal reservoirs, except molluscum, which is primarily a human disease. The reservoir is recognized and distributed globally in some cases, as is the case with ovine and bovine parapoxviruses. An occupational hazard of those who interact with the contaminated reservoir hosts is human contamination with these viruses.
Monkeypox is confined to West Africa, and squirrels are more critical than monkeys as reservoir hosts. The cowpox virus is limited to Europe and the western part of the former Soviet Union. Bovine cowpox is unusual, and the most frequently recorded host is the domestic cat. Lack of definitive knowledge on the cowpox virus reservoir host, but it is presumably tiny wild rodents. Cases occur without established contact with cats or cattle, and it is possible to spread indirectly through barbed wire or brambles.
Restricted natural person-to-person transmission of monkeypox, but not more than four or five generations, has been observed. Parapox and cowpox diseases spread from person to person occasionally, if ever. Molluscum’s person-to-person spread is historically associated with physical contact sports (e.g., wrestling) and towel sharing. However, there is growing evidence that molluscum sexual transmission is significant.
Traditionally, the Vaccinia virus is known as a laboratory virus without a natural reservoir. Although its host reservoir lacks information, the buffalopox virus, now considered a vaccine virus variant, appears to have established itself in India. It is important to note that such strains may become developed in animal populations and interact with genetically related viruses circulating in them due to the possible use of recombinant vaccinia virus vaccines.
Diagnosis:
In some instances, a right diagnosis would be made possible by the existence of the lesions and a careful history of interaction with the infected reservoir animal or other infected people; difficulties can occur if no such contact is known. This is perhaps most prominent with human cowpox, as most cases are not linked to a single source, and occasionally an anthrax clinical diagnosis is made.
An effective means of rapid diagnosis is electron microscopy of the vesicle or scab material; poxviruses and herpesviruses are readily differentiated, and the characteristic morphology of parapoxviruses can be identified. Immunofluorescence of infected cell cultures can distinguish morphologically related poxviruses from various genera for e.g., Orthopoxviruses and Yatapoxviruses. While the molluscum virus has yet to be cultivated in tissue culture and chicken embryos, the other poxviruses are easily isolated. Cultivation then enables biologic and serum neutralization samples to be established. Near antigenic relationships within genera compromise precise recognition through antibody detection, but knowledge of the host and geographic range will validate a presumptive diagnosis.
Treatment:
Infections of Variola have been globally eradicated. There is a concern, however, about the reintroduction of smallpox through bioterrorism. An international health care emergency would precipitate the reappearance of smallpox. Local and federal public health authorities should be informed of suspected cases of smallpox.
For smallpox or vaccinia, no known remedies are currently available. Several nucleoside and nucleotide analogues, including variola, vaccinia, monkeypox, cowpox, molluscum contagiosum, and orf, have shown Invitro activity and in vivo antiviral activity against various poxviruses. So far, Cidofovir and a number of its derivatives are the most efficient.